 الإهداءات
الإهداءات |
|
|
|||||||

| التسجيل | التعليمـــات | التقويم | مشاركات اليوم | البحث |
 |
|
|
أدوات الموضوع | انواع عرض الموضوع |
|
الإهداءات |
|
|
|||||||
| التسجيل | التعليمـــات | التقويم | مشاركات اليوم | البحث |
|
|
|
أدوات الموضوع | انواع عرض الموضوع |
|
#1
 05-09-2010, 11:57 PM
05-09-2010, 11:57 PM
|
||||||||
|
||||||||
|
المتلازمة - ليس مرضاً بحد ذاته، ولكن مجموعة من الأعراض المرضية يظهر أغلبها أو بعضاً منها قي حالات مرضية معينة، وعادة ما تسمى بإسم الشخص الذي قام بنشر بحث علمي عنها كمجموعة أعراض، وأغلب المتلازمات نتيجة عيوب في الصبغيات - الكروموسومات أو المورثات - الجينات

|
|
05-09-2010, 11:58 PM
|
#2 | |


|
الحثل العضلي الدوشيني مرض وراثي يصيب جميع أنواع العضلات في الجسم، ويتميز بضعف العضلات الذي يبدأ في عضلات الحوض، ثم يتطور بسرعة ليصيب جميع عضلات الجسم، وهو ما يؤدي إلى الأعاقة الحركية مبكراً ومن ثم الوفاة في منتصف العمر، ويبلغ معدل الإصابة واحد من 3500 ولادة من الذكورتقريباً، ونادراً ما يصيب البنات. Duchenne Muscular Dystrophy العضلات المتأثرة بالمرض ما هو السبب ؟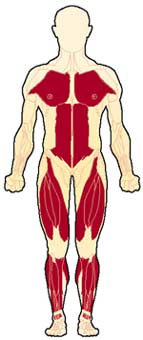 السبب مجهول ويتركز في وجود مورث غير طبيعي لمادة تسمى ديستروفين - dystrophin - وهي مادة بروتينية في العضلات، وينتقل هذا المورث عن طريق الكروموسوم الجنسي بالطريقة المتنحية X-linked recessive inherited، وهو ما يعني أن الأناث نادراً ما يصابون بالمرض لوجود زوج من الكروموسومات أحدهما طبيعي، ولكن الأم الحاملة للمرض لديها أحتمال ان تقوم بنقل المورث المعطوب لنصف أطفالها الذكور- ليكونوا مرضى، كما أن هناك أجتمالية ان تنقل المورث المعطوب لنصف بناتها - ليكونوا حاملين للمرض. هل يمكن أن تحدث الحالة في عائلة ليس لديها مصابين بالمرض؟ نعم - يمكن أن تحدث الحالة في عائلة ليس لديها مصابين بالمرض، وهو ما يسمى بالطفرة الجينية الوراثية new mutation، كما أن هناك احتمالية عدم ظهور المرض في الأجيال السابقة . أعراض المرض: المرض وراثي ، أي أنه موجود منذ الولادة، ولكن الأعراض عادة ما تبدأ في الظهور في سن الرابعة أو الخامسة من العمر، وتزداد تدريجياً وبسرعة كبيرة لتؤثر في جميع عضلات الجسم، وتتركز الأعراض في ما يلي: o ضعف العضلات o مشي غير طبيعي وكثير السقوط والتعثر o عدم القدرة على اداء النشاطات الحركية : صعوبة طلوع الدرج، الجري، القفز، اللعب o عدم القدرة على الإعتناء بالذات o عدم القدرة على المشي عند عمر 12 سنة o الاجهاد العام o انخفاض نسبة الذكاء - احتمالية o التشوهات الخلقية الجسمية: جنف العمود الفقري، التقفعات Contractures o العيوب العضلية: تضخم عضلة بطة الساق o المشاكل التنفسية: تكرر الإلتهابات الرؤية، الفشل الرئوي ، وهي من أهم اسباب الوفاة o تضخم عضلة القلب Cardiomyopathy، الهبوط القلب Congestive heart failure -نادرة الحدوث 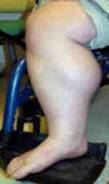 تضخم عضلة بطة الساق  ضعف العضلات 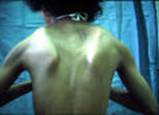 ضعف العضلات منظر من الظهر مجموعة صور توضح الطريقة المميزة لوقوف الطفل المصاب من الارض وتسمى علامة جاور Gower sign 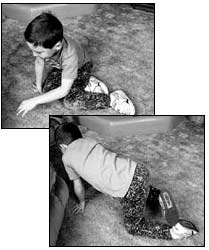  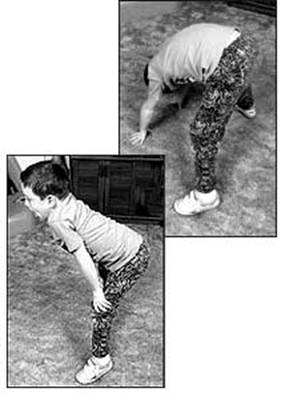 الحثل العضلي مرض متطور يزداد مع التقدم في العمر، نادراً ما تظهر الأعراض في السنة الأولى من العمر، وعادة ما تبدأ الأعراض قبل السادسة من العمر، تظهر على شكل ضعف متطور ومستمر وثابت في عضلات الرجلين والحوض، مصحوبة بضمور في العضلات، ويمتد الضعف ليصيب عضلات اليدين والكتفين والرقبة وبقية اجزاء الجسم ، وقد لوحظ ان شدة الإصابة في النصف السفلي أكثر منها في النصف العلوي من الجسم. يلاحظ ان عضلات باطن الساق تتضخم في البداية، وهو ما يسمى التضخم الكاذب، فالانسجة العضلية تضمر ويحل مكانها أنسجة دهنية و الانسجة الليفية connective tissue ، ضمور هذه العضلات يؤدي إلى تليفها وقصرها، وهو ما يؤدي إلى تفقعم الأطراف، ومن ثم إلى العوق الحركي للطفل، فيصبح محتاجاً للعكازات للمشي قبل بلوغه العاشرة من العمر، ويصبح المصاب يعتمد على الكرسي المتحرك قبل الثانية عشر من العمر ضعف العضلات والعيوب العظمية تؤثر على حركة الجهاز التنفسي، فيحدث تكرار للإلتهابات الرؤية، وهي ما قد يؤدي للوفاة في اوائل العشرينات من العمر التشخيص : يبدأ التشخيص مع ظهور الأعراض المميزة لهذا المرض، فضعف العضلات يبدأ من منطقة الحوض والرجلين حيث يكون هناك صعوبة في وقوف الطفل من وضع الجلوس، حيث يكون معتمداً على استخدام اليدين، ثم يتطور الضعف ليصيب بقية عضلات الجسم، كما أن هناك علامة مميزة أخرى وهي التورم الكاذب لعضلة بطة الساق، وقد يحتاج الطبيب المعالج لأجراء بعض الفحوص لتأكيد التشخيص ومنها: o تحليل كرياتينين فوسفوكاينيزserum CPK حيث يرتفع 50-100 مرة فوق المعدل الطبيعي o تخطيط العضلات الكهربائي electromyography، ويظهر ان الضعف في حركة العضلة ناتج من العضلات وليس من الأعصاب o فحص عينة من العضلة تحت المجهرmuscle biopsy، وتلك تثبت نوعية الخلل في العضلات o تخطيط القلب الكهربائي ، ويكشف وجود أي نشاط غير طبيعي للقلب العلاج: ليس هناك علاج يمنع المرض من الحدوث أو يزيله، ولكن يتم العلاج لتقليل تأثيرات المرض على الطفل المصاب، ومنها: o العلاج الطبيعي: والهدف منها تقليل التقفعات والعاهات وتاخير حدوثها، الحافظ على القوة العضلية، الحفاظ على اقصى جهد وظيفي، زيادة الحركة للمفاصل والوظيفة بواسطة الجبائر، الحفاظ على زيادة سعة التنفس o النشاط الرياضي: الخمول يساعد على زيادة الشد العضلي والتشوهات o المعالجة الجراحية: قد يفقد الطفل القدرة على المشي نتيجة تيبس العضلات والمفاصل، وغالباً ما يحدث ما بين 8-14 سنة، لذى فقد يحتاج للتدخل الجراحي لتحرير الشد والتشوه حول المفصل ليعطي مجال أوسع لحرية الحركة، كما قد يحتاج الطفل للجراحة عند زيادة حدة تقوس العمود الفقري o الأدوية: هناك بعض الأدوية المستخدمة لعلاج الشد العضلي o الدعم النفسي والاجتماعي.   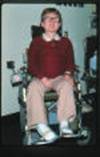 |
|
|
|
|
05-10-2010, 12:00 AM
|
#3 | |
|
متلازمة انجلمان Angelmans Syndrome الدكتور : عبدالله محمد الصبي - أخصائي طب الأطفال - مدينة الملك عبدالعزيز - الحرس الوطني  قام الطبيب الانجليزي انجلمان Harry Angelman بنشر بحث عام 1965 عن ثلاث حالات لا يوجد بينهم صفة القرابة ، أسماهم الأطفال الدمى Puppet Children ، تشترك في صفات محددة وهي : صفات وملامح متشابهة ، التخلف الفكري، نوبات أفراط في الضحك، نوبات الصرع، طريقة خاصة في المشي. .jpg) الأسباب: o الحالة غير وراثية ولكن طفرة جينية o حالة تعتبر نادرة ، ونسبة حدوثها حالة لكل 30.000 ولادة حية o ينتج بسبب قطع أو نقص في طرف الصبغي رقم 15 الجزء q11-q13 o نقص المورث UBE3A o غالباً ما يكون النقص في الكروموسوم المورث من الأم o اذا كان النقص في المورث من الأب فإنه يؤدي لمتلازمة برادر ويلي Prader-Willi Syndrome، ولكن أكتشف أختلاف بسيط في المكان للمورث .jpg) التشخيص: o الأعراض المرضية والتي يمكن ملاحظتها بين عمر الثالثة والسابعة من العمر o تحليل الكروموسومات o تقنية التهجين الفلوري للكروموسوم FISH o أختبار دي-أن-أي DNA وأختبار المورث الخاص UBE3A o الأشعة المقطعية CT brain وارنين المغناطيسي للدماغ MRI طبيعي o التحاليل الكيماوية للدم طبيعية .jpg) الأعراض المرضية : o وزن الطفل وملامحه طبيعية عند الولادة o تأخر في اكتساب المهارات الفكرية والحركية ، مع عدم فقد أي مهارة مكتسبة o صعوبة في الرضاعة مع كثرة التقيؤ o تظهر الملامح الجسدية والسلوكيات المرضية ما بين الثالثة والسادسة من العمر o التخلف الفكري المتوسط إلى الشديد o تأخر اللغة والتخاطب مع ضعف التواصل o نوبات أفراط في الضحك - لذلك سمي الدمية الضاحكة o نوبات الصرع o طريقة خاصة في المشي .jpg) الصفات الجسمية: o ملامحه طبيعية عند الولادة o صغر حجم الرأس بعد عمر السنتين o العينان حادتان o فم واسع مبتسم o سيلان اللعاب، خروج اللسان o الجزء الأوسط من الوجه ناقص النمو o نتوء الاسنان o الحول o في مرحلة الشباب يستطيل الوجه وتصبح الصفات أكثر وضوحاً o حساسية من ارتفاع حرارة الجو .jpg) نوبات أفراط في الضحك : o نوبات متكررة ومفرطة o سرعة الاستثارة o تصفيق باليدين o فرط الحركة .jpg) نوبات الصرع : o تظهر النوبات الصرعية في الطفولة المبكرة - السنة الثالثة - وعادة ما تختفي مع البلوغ o يمكن حدوث نوع أو أكثر من أنواع الصرع : الصرع العام، الصرع الجزئي، الشرود المفاجيء o تؤثر على 80% من الحالات o تظهر على شكل نوبات من الارتعاش o عادة يتم السيطرة عليها بأدوية الصرع o رسم المخ EEG غير طبيعي o الصرع ينتج نتيجة نقص مادة GABA ، والمورث الخاص بها موجود على الكروموسوم رقم 15، هذه المادة كابحة لنشاط المخ .jpg) المشي والحركة والإتزان : o حركات غير طبيعية للجذع والأطراف يمكن ملاحظتها منذ السنة الأولى من العمر o الحركات الإرادية غير متزنة o عدم الإتزان في المشي o المشي على أطراف الأصابع o في بعض الأطفال ، زيادة الحركات غير الارادية قد تمنعهم من الحبو والحركة والمشي .jpg) العلاج : o أدوية الصرع مثل الفالبوريت valproic acid (e.g.,Depakote)، التوبامكس topiramate (Topamax)، التجراتول carbamazepine (Tegretol)، كلونازيبام clonazepam (Klonopin) o الحمية الغذائية ketogenic diet o برنامج تعديل السلوك o برامج اللغة والتخاطب o الخدمات التأهيلية .jpg)
|
|
|
|
|
05-10-2010, 12:01 AM
|
#4 | |
|
متلازمة روبينشتاين - تايبي الدكتور : عبدالله محمد الصبي - أخصائي طب الأطفال - مدينة الملك عبدالعزيز - الحرس الوطنيRubinstein-Taybi Syndrome متلازمة الإبهام الكبير  تم التعرف على هذه المتلازمة عندما قام الطبيبان جاك روبينشتاين وهوشانغ تايبي Dr. Jack Rubinstein and Dr. Hooshang Taybi بنشر دراستهم عام 1963، حيث وجدوا تشابه في الصفات الجسمية وتأخر في التطور لدى عدد من الأطفال غير الأقرباء، هناك قصر القمة، تأخر النمو الفكري والحركي، سمات الوجه، كبر حجم الأصبع الكبير والسبابة في اليدين والقدمين، تأخر فكري، وقد توالى بعد ذالك تسجيل تلك الحالات من كل أنحاء العالم الأسباب : o الأسباب غير معروفة o تعتبر طفرة جينية في أغلب الحالات، حالة غير وراثية o نسبة حدوثه حالة لكل 300.000 مولود o يصيب الذكور والإناث بنفس النسبة تقريباً o نقص في الذراع القصير للكروموسوم رقم 16(16p13.3) o طفرة في المورث - الجين CREBB o أحتمالية تكرار الحالة لنفس العائلة 1 لكل ألف o أحتماية حدوث الحالة لأولاد المصاب / المصابة 50% - الوراثة السائدة autosomal dominant .jpg) التشخيص: o عادة ما يتم التشخيص في نهاية السنة الثانية من العمر أو بعد ذلك o العلامات الجسمية o فحص الكرموسومات ، التهجين الفلوري الموضعي FISH يظهر العيوب في الكرموسوم رقم 16 o طفرة في المورث CREBB o تخطيط المخ EEG غير طبيعي في أغلب الحالات o الأشعة المقطعية للرأس تظهر أتساع في ثقب قاع الجمجمة agenesis of corpus callosum, large foramen magnum .jpg) الأعراض المرضية : o التخلف الفكري - معدل الذكاء 30-79 o نقص القدرة على التركيز o تأخر النمو الحركي o الصفات الجسمية، الصفات المميزة لليدين والقدمين o مشية متصلبة غير ثابتة o نوبات صرع في ربع الحالات o تأخر النطق والتخاطب، ونسبة عالية تستخدم لغة الاشارة o عادة ما يكون الأطفال مسالمين ودودين أجتماعيين .jpg) الصفات الجسمية: o قصر القامة o تأخر النمو العظمي o صغر حجم الرأس، بروز الجبهة o الشعر كثيف o العينان مائلتان إلى الخارج وإلى الأسفل o وجود زوائد في زاوية العين الداخلية o ارتخاء الجفن العلوي o بروز الأنف o الفم ضيق o ارتفاع سقف الفم o عدم انتظام الأسنان o أنحناء جانبي للعمود الفقري مع تشوه الفقرات .jpg) .jpg) .jpg) اليدين والقدمين: o من الصفات الاساسية لهذه المتلازمة o كبر حجم الأصبع الكبير في اليدين والقدمين مع أنحناء إلى الجهة الداخلية o قد تكون جميع أصابع اليدين والقدمين كبيرة مع وجود أظافر مسطحة وعريضة .jpg) .jpg) .jpg) مشاكل مرضية مصاحبة: o الحول o الماء الأزرق وقد يؤدي للعمي o نقص السمع 25% من الحالات o تكرار التهاب الجهاز التنفسي العلوي والتهابات الأذن o عيوب خلقية في القلب والكلى في 35-40% من الحالات .jpg) العلاج : o لا يوجد علاج لهذه الحالات ، فعيوب الكرموسومات لم يكتشف علاج لها o الرعاية الصحية o الخدمات التأهلية o علاج الصرع o علاج النطق والتخاطب .jpg) .jpg) |
|
|
|
|
05-10-2010, 12:02 AM
|
#5 | |
|
متلازمة كورنيلا دي لانج الدكتور : عبدالله محمد الصبي - أخصائي طب الأطفال - مدينة الملك عبدالعزيز - الحرس الوطنيCornelia de Lange syndrome (CdLS) متلازمة أقزام أمستردام Amsterdam Dwarf Syndrome  وصف الدكتور براشمان عام 1916م مجموعة من الحالات تتميز بالشكل المميز للرأس والأطراف وقصر القامة، وعرفت بأقزام أمستردام، ثم تبعه مواطنه الهولندي طبيب الأطفال الدكتور كورنيلا دي لانج عام 1933، والذي سميت هذه المتلازمة بإسمه، وكلاهما قام بالشرح عن صفات الحالة ومميزاتها التشخيصية. الأسباب : o السبب غير معروف o نسبة حدوثها حالة لكل 10.000- 30.000 ولادة حية o تصيب الأولاد والبنات بنفس النسبة o تصيب كل الأعراق والمجتمعات o أغلب الحالات تنتج عن طفرة وراثية، لعدم وجود مثل تلك الحالة في العائلة o يعتقد أنها تنتقل عن طريق الوراثة السائدة - وهو ما يعني أن عطباً في أحد الجينات يمكن أن يظهر الأعراض كاملة o يعتقد أن السبب في حدوث الحالة هو عطب في المورث NIPBL gene، الذي يقوم بصناعة بروتين يسمى delangin، وهذا المورث موجود على الكروموسوم رقم chromosome 3 band q26-27 o اذا كان أحد الوالدين مصاب اصابة خفيفة ، فحسب الوراثة السائدة فإن أحتمالية ولادة طفل مصاب هي 50% o اذا لم يكن هناك طفل مصاب في العائلة، فإن نسبة التكرار هي 0.5-1.5 من الولادات اللاحقة فقط. العلامات المميزة للحالة : هناك العديد من العلامات الجسمية والتغيرات المصاحبة للحالة، ولكن ليس من المفترض وجود جميع تلك العلامات في كل الأطفال المصابين بالحالة، وفي ما يلي سنوجز العلامات المتكررة في أغلب الحالات ومنها: o نقص الوزن عند الولادة - أقل من ثلاثة كجم o البكاء بطريقة ضعيفة لدى المواليد o صعوبة التنفس والرضاعة لدى المواليد o زيادة توتر العضلات o قصر القامة o ضعف النمو o ضعف التطور الحركي والفكري o صعوبات في التغذية o علامات مميزة للرأس والأطراف o ضعف السمع o مشاكل بصرية : رأرأة العين، بعد النضر، قصر النضر o الترجيع المعدي o التشنج والصرع o عيوب خلقية في القلب o تخلف فكري - بسيط إلى المتوسط o تأخر النطق والتخاطب o عدم نزول الخصية .jpg) .jpg) .jpg) العلامات المميزة للرأس : o صغر حجم ومحيط الرأس o صغر حجم الحاجب، والتقاء الحاجبين سوياً o زيادة طول الرموش o كثافة شعر الرأس o قصر الرقبة o الأنف صغير o الشفة العليا صغيرة ومرتفعة للأعلى o الأسنان صغيرة ومتباعدة o الأذن صغيرة ومنخفضة o شق الحنك .jpg) العلامات المميزة للأطراف : o أختلاف حجم الأطراف، وقد يكون هناك طرف ناقص o صغر حجم اليدين والقدمين o التحام جزئي لأصابع القدمين الثاني والثالث o الأصبع الخامس في اليدين معوج o زيادة كمية الشعر في الجسم .jpg) .jpg) .jpg) تأخر النمو : o نقص الوزن عند الولادة o قصر القامة o ضعف النمو، ويكون سرعة النمو خاصة بهم o عادة ما يصلون للبلوغ كأقرانهم o عند البلوغ يكونون قصار القامة .jpg) التطور الحركي والفكري : هناك أختلاف كبير بين المصابين بهذه الحالة في التطور الحركي والفكري، فالبعض لديهم تأخر شديد في حين نرى البعض الآخر بنسبة ذكاء طبيعية، والمصابين بتأخر شيديد نلاحظ لديهم تأخر في النطق والتخاطب، أما الحالات المتوسطة فيمكنهم الكلام في حوالي الخامسة من العمر، وعادة ما يستطيعون الحركة والمشي بعد سن الثانية. .jpg) المشاكل السلوكية : هناك العديد من المشاكل، ومنها : o أيذاء الذات o عدم الاحساس بالارتياح والتضايق o بعض العلامات التوحدية، الانعزالية ، عدم التواصل مع الآخرين، الحركات المتكررة o عدم التفاعل مع الألم o النشط الزائد o صعوبة في النوم .jpg) التشخيص : o لا يوجد أختبارات أو تحاليل للتشخيص o التشخيص يعتمد على الأعراض والمميزات الجسمية التحاليل : هي الفحوص التي يمكن عملها للطفل لمتابعة الحالة وليست لتشخيص المتلازمة، ومنها: o صورة الكروموسومات o أشعة للعظام - تأخر نمو العظام o صورة الدم - نقص عدد الصفائح الدموية o الاشعة الصوتية للقلب o قياس السمع o كشف البصر والعيون o قياس هرمون النمو growth hormone .jpg) العلاج : o لا يوجد علاج شاف للحالة o التدخل المبكر، العلاج الكلامي، العلاج الوظيفي، العلاج الطبيعي ، يمكنها تقليل الأعراض المصاحبة المستقبل للطفل : المستقبل علمه عند الله ، ولكن مع التدخل العلاجي للأعراض المصاحبة فيمكن للطفل العيش لمدة طويلة ، وتختلف الحالات، فالبعض يعيش معتمداً على نفسه، والآخرين قد يعتمدون أعتمادا كليا في حياتهم على الآخرين. .jpg)
|
|
|
|
|
05-10-2010, 12:04 AM
|
#6 | |
|
متلازمة ويليامز الدكتور : عبدالله محمد الصبي - أخصائي طب الأطفال - مدينة الملك عبدالعزيز - الحرس الوطنيWilliam`s Syndrome  .jpg) .jpg) .jpg) تم أكتشاف هذه الحالات الطبيب الايرلندي ويليامز عام 1961، حيث لاحظ التشابهة بين أربعة أطفال ليس بينهم قرابة، حيث ملامح الوجه مميزة، مع وجود عوق فكري، ارتفاع نسبة الكالسيوم في الدم، تضيق في مجرى الشريان الأبهر للقلب- الاورطي .jpg) ما هي نسبة حدوث الحالة: o حالة واحدة لكل 000 20 ولادة o يصيب الذكور والإناث o جميع مناطق ودول العالم بنفس النسبة تقريباً o ليس هناك زيادة في تكرار الحالة مع وجود طفل مصاب لدى العائلة o المصاب لديه أحتمالية 50% لينقل المورث لأبناءه وبناته الأسباب: o السبب غير معروف، الحالات فردية ، غير وراثية o طفرة جينية - ليس السبب شيئاً فعله الوالدين أو لم يفعلوه خلال الحمل أو قبله o حالات قليلة تكون وراثية ، أي تنتقل من الوالدين للطفل o نقص أو حذف جزء من الكرموسوم - الصبغي - السابع q11 ، وهناك أكثر من 15 مورث في هذا الجزء o في هذا الجزء مورث مادة elastin، لذلك يلاحظ ارتخاء العضلات، الفتق، ارتخاء الشريان الأبهر o في هذا الجزء مورث يدعى ليم LIM-kinase I، والذي ينشط في الدماغ، وهذا يشير إلى إمكانية تأثيره في نمو الدماغ وقيامه بوظائفه .jpg) .jpg) التشخيص: o علامات الحالة قد لا تكون واضحة، وقد تمر سنوات عديدة قبل القيام بتشخيصها o لا يعتمد في التشخيص على العلامات الظاهرة بل يحتاج إلى اختبارات خاصة o يتم التشخيص من قبل طبيب الأطفال وطبيب الأمراض الوراثية o يتم التشخيص عن طريق فحص الكروموسومات، وفحص التهجين الفلوري الموضعي ( Fluorescent in situ hybridization ( FISH، ويكون هذا الفحص إيجابي (غير طبيعي)في حوالي 95 % من الأطفال المصابين بمتلازمة وليامز o زيادة التكلس في العظام، العمود الفقري، قاعدة الدماغ .jpg) الأعراض المرضية : العلامات الرئيسة العامة لمتلازمة ويليامز هي : o نقص الوزن عند الولادة، قلة النمو بعد الولادة o ملامح الوجه مميزة o التخلف الفكري البسيط الى المتوسط o صعوبة النطق والكلام o زيادة نسبة الكالسيوم في الدم o مشاكل القلب والجهاز الدوري o مشاكل التغذية o المغص المتكرر في المواليد o مشاكل الأسنان : قد تغيب بعض الاسنان بشكل كامل، الاسنان ناقصة التصنع o عيوب الكلى: تشوهات كلوية مثل تكلس الكلية، عدم تناظر في حجم الكليتين، كلية وحيدة ومتوضعة في الحوض، رتج مثاني وتضيق في الاحليل، جذر مثاني حالبي o الفتاق: فتق السرة وفتق اربي o فرط حساسية للصوت o مشاكل الجهاز الحركي - العظام والعضلات o التكيف الاجتماعي والمشاكل النفسية o ضعف النمو والتطور الحركي والفكري، صعوبات التعلم، ضعف التركيز o سجلت حالات قليلة من الوفيات اثر التخدير العام .jpg) ملامح الوجه مميزة : يمكن القول أن أغلب الأطفال المصابين يتشابهون في ملامح الوجه المميزة، ولكن في الكثير من الحالات لا يمكن تمييزها ومعرفتها ، وتحتاج إلى متخصص، فالأطفال عادة ما يشبهون والديهم في الكثير من السمات، وعادة ما تزداد هذه العلامات مع التقدم في العمر، ومن هذه العلامات: o صغر حجم الرأس عند الولادة microcephaly o الشكل المميز للوجه elfin o الأنف صغير، انخفاض قاعدة الانف o الشق الطولي اعلى الشفة العلوية يكون طويلا long philtrum o الفم المفتوح والشفاه الغليضة o الذقن صغير o تورم حول العينين ، طية جانب العين من جهة الانف o العينين زرقاء أو خضراء o عدم التحكم في عضلات الوجه والفم .jpg) مشاكل القلب والجهاز الدوري : التشوهات القلبية موجودة غالبا لدى المصابين بمتلازمة ويليامز ، واهمها : o عيوب في صمامات القلب ( ضيق المنطقة التي فوق الصمام الأورطي- الأبهر) supravalvular aortic stenos o تضيق الشريان الرئوي pulmonary arteries o عيوب خلقية في الفتحات ما بين الاذينتين وما بين البطينين o بشكل اقل قد يكون هناك تضيق في الشريان الكلوي و ارتفاع في التوتر الشرياني .jpg) زيادة نسبة الكالسيوم في الدم : لوحظ أن نسبة من الأطفال المصابين بمتلازمة ويليامز لديهم أرتفاع في نسبة الكالسيوم في الدم، ولكن نسبة حدوثه غير معروفة، كما أن الاسباب لحدوث هذه الزيادة غير معروفه، تلاحظ هذه الزيادة بعد السنة الثانية من العمر، هذه الزيادة في نسبة الكالسيوم في الدم ممكن أن تؤدي إلى المغص المتكرر وآلام البطن، وقد يحتاج الطفل إلى العلاج الغذائي والدوائي، وفي أغلب الحالات يختفي هذا الارتفاع من تلقاء نفسه، ولكن يجب متابعة الحالة. نقص النمو والتغذية : يلاحظ أن الكثير من المواليد يكونوا ناقصي النمو عند الولادة مقارنة بأخواتهم، كما يلاحظ نقص الزيادة في الوزن في السنوات الأولى من العمر، مما قد يستدعي البحث عن أسباب نقص الوزن والنمو، كما يلاحظ ضعف وصعوبة التغذية قد تعزى لضعف العضلات ، ولكن عادة ما تتحسن مع مرور السنوات. .jpg) الجهاز الحركي: o حركة المفاصل غير كاملة o نقص في تصنع الأظافر o تشوهات العمود الفقري و اهمها الحدب والجنف o المنعكسات الوترية زائدة النشاط o شلل الحنجرة o نقص تصنع الرحى و التحام عظم الكعبرة مع عظم الزند .jpg) التطور الفكري - الاجتماعي - الحركي : أغلب الأطفال المصابين بمتلازمة ويليامز لديهم درجة من درجات التخلف الفكري والحركي، وأن كان هناك تشلبه بينهم فهناك اختلاف كذلك ، ومن أهم العلامات لديهم: o مستوى الذكاء متوسط o مشاكل التحصيل الدراسي o تتركز صعوبات التعلم لديهم في مجال القراءة والكتابة والحساب o ضعف التركيز o ضعف خفيف في التكيف الاجتماعي o الوظيفة الادراكية والحركية متأثرة اكثر من الكلام و الذاكرة و اللغة o تأخر المشي، الكلام، التدريب على الحمام o مسالمين، شخصيتهم ودودة و مهذّبة، حسن المخالطة، وكثرة الحديث. o ومتميزين وبشكل واضح بعض المجالات، في التحدث، التعرف على الوجوه o وفرة من المفردات اللغوية - قد تضلل المعلمين فتجعلهم يعتقدون أن لدى هؤلاء الأطفال مهارات فكرية o موهبة موسيقية فذة، فيصغون إلى الموسيقى ويغنون ويعزفون على الآلات بشكل مدهش .jpg) العلاج: o حتى الآن لا يوجد علاج لتعديل النقص في الكروموسوم o المتابعة الطبية للأعراض المصاحبة مثل أمراض القلب والكلى وغيرها o قياس ضغط الدم بشكل دوري o قياس مستوى الكالسيوم بشكل دوري o يجب التركيز على صقل مهارات الطفل التعليمية و تدريبه و تعليمه .jpg) المستقبل: - العلم عند الله o عادة ما يقوم البالغين بالإعتماد على أنفسهم في الكثير من أمور الحياة o صعوبات التعلم - يحتاج إلى التعليم الخاص o يمكنهم العمل تحت أشراف خاص .jpg)
|
|
|
|
|
05-10-2010, 12:06 AM
|
#7 | |
|
متلازمة مارافان
Marfan's Syndrome الدكتور : عبدالله محمد الصبي - أخصائي طب الأطفال - مدينة الملك عبدالعزيز - الحرس الوطني مرض وراثي ينتقل من جيل لأخر عن طريق ما يسمى بالوراثة السائدة autosomal dominant trait، أول من قام بوصفها ونشرها الدكتور مارفان Dr. Antoine Marfan عام 1896م، وهي حالة تحدث نتيجة خلل في تركيبة النسيج الضام - Connective Tissue- والذي يدخل في تركيبة العديد من أعضاء الجسم، مما يؤدي إلى ظهور العديد من العيوب في العظام، الجهاز الدوري، العين، وغيرها، كما يؤدي إلى صعوبة في التعلم. .jpg) نسبة الانتشار والأسباب : o حالة نادرة تصيب فرد لكل 10.000 o يصيب الذكور والإناث بنفس النسبة o تحدث الأعراض نتيجة لخلل في مورث- جين- يسمى بمورث الفيبريلين رقم واحد(Fibrillin 1) الموجود على الذراع الطويلة لكروموسوم chromosome 15q21.1. o تدخل هذه المادة في تركيب النسيج الضام ، ومن ثم في تركيبة العديد من أعضاء الجسم كالأوعية الدموية، صمامات القلب،العظام، الجلد، والعين . o مرض وراثي ينتقل من جيل لأخر عن طريق ما يسمى بالوراثة السائدة autosomal dominant trait o اذا كان أحد الوالدين مصاب - هناك أحتمالية أنتقال الحالة لأطفاله 50% o اذا كان أحد الوالدين مصاب، ولديهم طفل مصاب، فإن احتمالية تكرار الحالة 50% من المواليد o في حوالي 25 بالمائة من الحالات يحدث لدى عائلة ليس فيها مصاب وهو ما يسمى بالطفرة الوراثية Mutation الأعراض : تختلف الأعراض المرضية بين شخص وآخر، كما تختلف بين أفراد العائلة الواحدة المصابين بالحالة، فقد نجد لدى البعض عرض أو أثنين، وقد تكون شديدة لدى الآخرين، ومن أهم الأعراض : o طول القامة والأطراف o الشكل المميز للرأس والوجه o التغيرات في العمود الفقري والصدر o مشاكل العين o مشاكل في الجهاز الدوري والقلب o كثرة حدوث الفتوق الاربية والفخذية o خلوعٍ المفصل المتكرر o تخلف عقلي بسيط في بعض الحالات o صعوبة بالتعلم في بعض الحالات غير المصابة بتخلف عقلي 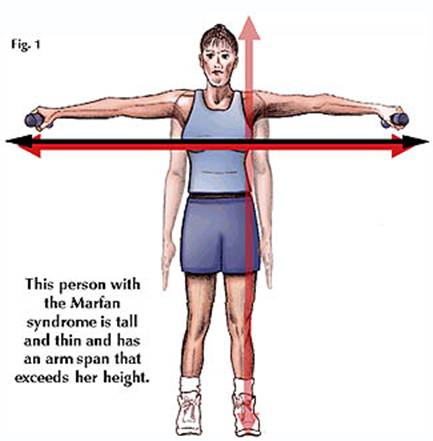 الشكل العام والأطراف : o طول القامة نتيجة لطول عظام الأطراف - أطول من المعدل الطبيعي نسبة لعمرهم ولباقي أفراد أسرتهم o الأطراف العليا والسفلي نحيفة و طويلة o يكون قيـاس باعه - البعد بين الذراعين - أكبر من طول الجسم o يزيد قياس القدم الى العانة عن البعد بين العانة وقمة الرأس o طول ملحوظ في أصابع اليدين والقدمين o تكون الأصابع طويلة دقيقة مفرطة البسط - عنكبية الأصابع Arachnodactyly o علامة المعصم o تكون عضلات الهيكل ضعيفة النمو وناقصة التوتر o يؤدي ضعف الأربطة إلى الحركة المضاعفة للمفصل أو رخاوته .jpg) .jpg) .jpg) .jpg) .jpg) الرأس والوجه: o يبدو رأس طويل مع صغر حجم الوجه o بروز عظام جبهة الرأس o ارتفاع في سقف الحلق o تكون الأسنان متزاحمة ومتراكبة o يكون الحنك مرتفعاً ومتقوساً العمود الفقري والصدر o الصدر الغاطس: عظمة القص - الصدر- غائر - صدر مقعر بشكل واضح، يجعل الصدر يأخذ شكل صدر الحمامة .jpg) o الصدر الناطح: عظمة القص - الصدر- بارزة إلى الأمام بشكل واضح - قمعي .jpg) o الظهر والعمود الفقري قد يزيد تقوسه - الحدب الجنفي .jpg) الجهاز الدوري- مشاكل القلب والشريان الأبهر : المشاكل القلبية وأصابات الشريان الأبهر - الأورطى - تؤثر في 80-90% من المصابين بمتلازمة مارفان، وهي السبب الرئيسي في أغلب حالات الوفاة، ومن أهم المشاكل: o ارتخاء الصمام الميترالي o ترجيع الصمام الميترالي o توسع جذر الأبهر (الأورطي) o ترجيع الصمام الأبهر o توسع و تورم دموي في جذر الأبهر Aneurysm .jpg) .gif) الرئتين : تكون الألياف الضامة جدران الأكياس الهوائية في الرئة، ووجود الضعف فيها يجعلها غير قابلة للتمدد والانقباض، مما قد يؤدي إلى التمدد الزائد أو تكرار الانفجار، مما يؤدي إلى وجود هواء خارج الأكياس الرئوية pneumothorax العلامات السلوكية والعقلية: o تخلف عقلي بسيط o صعوبة بالتعليم في بعض الحالات غير المصابة بتخلف عقلي o المشاكل السلوكية والنفسية والاجتماعية المصاحبة مشاكل العين : تظهر مشاكل العين في نصف المصابين تقريباً، فقد يؤدي ضعف الأنسجة الداعمة لعدسة العين إلى خلعها الجزئي، وغالباً للاتجاه نحو الأعلى ونحو الأمام، وهناك مشاكل في العين والنظر منها: o تسطح القرنية o قصر النضر o ازرقاق في بياض العين o عيوب العدسات o أنفصال الشبكية o الماء الأبيض - الساد o الماء الأزرق - الجلوكوما .jpg) .jpg) التشخيص : - ليس كل فرد طويل أو أطرافه طويلة مصاب بهذه المتلازمة، بل قد يكون الطول طبيعياً متوارثاً في العائلة - ليس هناك تحليل في المختبرللتشخيص - لذى فإن الطبيب المتخصص يمكنه الوصول للتشخيص من خلال ما أتفق عليه المتخصصون من قواعد للتشخيص المسماة Ghent criteria، وذلك بعد القيام بما يلي : o تاريخ العائلة الطبي o هل يوجد في العائلة أفراد طوال القامة o هل يوجد في العائلة مصابون بالامراض القلبية o الفحص الطبي السريري o الكشف بالأشعة الصوتية التشخيص الفارق والحالات المشابهة: ليس كل حالات طول القامة هي حالات متلازمة مارفان، فهناك الحالات العائلية الطبيعية، كما أن هناك حالات أخرى منها: o متلازمة أهلر - دانلوس Ehlers-Danlos Syndrome o متلازمة الكروموسوم الجنسي الهش Fragile X Syndrome o متلازمة كلاينفلتر Klinefelter Syndrome o مرض الضخامة Gigantism and Acromegaly o ارتفاع هرمون الغدة النخامية Hyperpituitarism o ارتفاع هرمون الغدة الدرقية Hyperthyroidism العلاج والعناية العامة : o عناية صحية مستمرة o المتابعة الدورية مع طبيب مختص في أمراض القلب، وقد يحتاج الأمر إلى إجراء الأشعة الصوتية للقلب كل ستة أشهر o عناية تعليمية خاصة للمتخلفين عقليا ومن لديهم صعوبات تعلم o عناية خاصة بالعين ومشاكل رؤية o الجراحة : قد تحتاج بعض الحالات إلى التدخل الجراحي المبكر مثل حالات توسع الشريان الأبهر وغيره o تجنب الأنشطة الرياضية التي يصاحبها أحتكاك وتصادم ، مما يمكن أن تؤديه من مشاكل قلبية أو أضرار بالعين ما هو مآل الحالة ؟ متلازمة مارفان مستمرة طوال العمر، تختلف الأعراض لكل مرحلة عمرية، كما تختلف من شخص لآخر حسب درجة تأثيراتها، ومع المتابعة المستمرة، والتدخل العلاجي والجراحي فإن النتائج جيدة. |
|
|
|
|
05-10-2010, 12:09 AM
|
#8 | |
|
متلازمة برادر- ويلي Prader-Willi syndrome الدكتور : عبدالله محمد الصبي – أخصائي طب الأطفال – مدينة الملك عبدالعزيز – الحرس الوطني عرفت هذه المتلازمة عندما قام أخصائيو الغدد برادر – لابهارت – ويلي بنشر تقرير عام 1956 في المؤتمر الدولي الثامن لطب الأطفال في كوبنهاجن، عن حالات فيها صفات متعددة مثل : إعاقة عقلية ، الرخاوة، مشاكل في التغذية، السمنة، عدم اكتمال الاجهزة الجنسية، القصر، تاخر النمو، وغيرها، وفي الستينات من القرن الماضي تمت دراسات أخرى وتمت أضافة العديد من الاعراض، كما تمت أضافة الاعراض السلوكية في السبعينات، وقد أثبتت الدراسات أنه مع التحكم في النظام الغذائي فإن هذه الحالات يمكن أن تعيش لمدة أطول، وقد تم تتبع العديد من الحالات الذين بلغوا العشرينيات من العمر. الانتشار : o تعد متلازمة برادر ويلي من الاضطرابات الجينية النادرة إذ يقدر حدوثها حالة لكل 12.000-15.000 ولادة o تتساوى نسبة الإصابة بين كل من الذكور والإناث o كل الأعراق o أغلب الحالات غير وراثية، ولكنها تحدث كطفرة جينية لأسباب غير معروفة o في نسبة قليلة لا تتجاوز 2% وجد لها أسباب وراثية وترتفع نسبة تكرار الحالة لدى العائلة o أحتمالية التكرار ضعيفة ماعدا الحالات النادرة وهي imprinting mutations, translocations, or inversions الأسباب: تنتج متلازمة برادر ويلي في معظم حالاتها من غياب أو حذف لجين من الذراع الطويل لكروموسوم 15 القادم من الأب ويرمز لها 15q11-q13، وفي بعض الأحيان تنتج من حصول الطفل المصاب على نسختين من الكروموسوم 15 من الأم، وهنا لا بد من توضيح الأمر : o السبب في النقص في طرف الكروموسوم رقم 15 غير معروف o وجد أن النقص يكون في الكروموسوم المنتقل من الأب بدون أسباب وراثية في 70% من الحالات o في 25% من الحالات وجد أن هناك الزوج من الكروموسوم رقم 15 من الأم بدون وجود كروموسوم الأب o في 2-5% من الحالات وجد أن النسخة رقم 15 من الأب في الكروموسوم لا تعمل. .gif) الخصائص والأعراض المرضية : o نقص الحركة وهو جنين . o ضعف العضلات منذ الطفولة ويزيد مع العمر o صعوبة في الرضاعة o خمول في مرحلة الطفولة مع بكاء ضعيف. o تأخر عام في النمو الحركي والفكري o ملامح جسمية مميزة o نقص وعدم زيادة في الوزن في مرحلة الطفولة. o شراهة في الأكل، بدانة بمرور الوقت، زيادة أو سرعة في الوزن مع سمنة مفرطة في غياب التدخل العلاجي . o مشكلات في الأكل ، مثل الارتباط الزائد بطعام معين والأكل الزائد. o تخلف عقلي بسيط أو صعوبات تعلم . o القصور الجنسي والأعضاء التناسلية o مشاكل سلوكية o الوفاة المبكرة بسبب زيادة الوزن .jpg) .jpg) الأعراض الجسمية : o ملامح جسمية مميزة o رأس صغير o استطالة الجمجمة مع وجه ضيق وعينين ضيقتين وبيضاويتين o أنخفاض الفم o شعر أشقر وعيون زرقاء مهما كانت الجنسية في 50% من الحالات o قصر القامة، قصر الأيدي والأقدام .jpg) ليونة العضلات : من العلامات المميزة لمتلازمة برادر ولي، ويبدأ من الولادة ويتحسن تدريجياً مع التقدم في العمر، ومن علاماته في مرحلة الطفولة ضعف التغذية وعدم المقدرة على المص ، ضعف البكاء، ضعف الحركة وتأخر التطور الحركي، كما يلاحظ عجز في التنسيق الحركي، كسل العين – الحول. .jpg) العوق الفكري: من العلامات المميزة غير معروفة السبب ، حيث لوحظ أن أكثر 95% من الحالات لديهم درجة من درجات التخلف الفكري، وفي الغالب يكون معدل الذكاء بين 70-79 ، ولكن يمكن أن يكون أقل من ذلك، أو يكون في المعدل الطبيعي. الأطفال الذين لديهم معدل ذكاء طبيعي لوحظ لديهم صعوبات في التعلم، ضعف التركيز والإنتباه، ولكن لديهم نقاط قوة مثل الذاكرة ، القدرة على القراءة، وغيرها. النطق والكلام : ارتخاء العضلات قد يؤدي لصعوبات في التغذية، كما يؤدي إلى ضعف حركات الفم ومن ثم تأخر النطق والكلام، وهو ما قد يعني الاحتياج للتدريب على النطق والكلام ، وقد يحتاج الأمر إلى التواصل عن طريق لغة الإشارة، وعادة ما تتحسن اللغة لدى هؤلاء الأطفال، وتكون لغتهم جيدة. ضعف النمو : ضعف النمو يبدأ من الولادة، حيث يلاحظ ضعف الرضاعة، مما قد يستدعي التغذية عن طريق الأنبوب الفمي المعدي، وهنا يجب ملاحظة أحتياج الطفل من الغذاء كما الزيادة الطبيعية للوزن. عادة ما يكون هرمون النمو ناقصاً لدى هؤلاء الأطفال، وهو ما يؤدي لقصر القامة، كما زيادة نسبة الدهون، ويحتاج الطفل إلى تعويض الهرمون الناقص في مرحلة الطفولة كما البالغين. .jpg) البدانة والشهية الشرهة: الشراهة في الأكل سببها مركزي في الدماغ، لعدم عمل الأجزاء الخاصة بالشبع، قد لا تكون ملاحظة في السنوات الأولى من العمر لضعف عضلات المص والبلع، ولكنها تزداد مع التقدم في العمر. عدم التحكم في كمية الطعام وضعف حركة الجسم تؤدي إلى السمنة، وتظهر البدانة عادة بعد سن الثانية أو الثالثة من العمر، لذى يجب أن يتناول المصابين كمية سعرات حرارية أقل من أقرانهم لمنع حدوث البدانة، وهو من الاسباب الرئيسة للوفاة، وحتى الآن لم يكتشف دواء قادر على ايقاف الشراهة في الأكل، وحتى الجراحة لا تجدي في تلك الحالات. المشاكل السلوكية: عادة ما يكون هؤلاء الأطفال طبيعيين هادئين في المراحل الأولى قبل سن المدرسة، ويحدث التغيير بعد ذلك، حيث يلاحظ عليهم تكرر حالات الغضب- الأنفعال- الأنطواء – الروتين، وتلك المشاكل غير مرتبطة بالسلوكيات الغذائية، ولكن التغيرات الغذائية يمكن أن تزيد حدتها، وعادة ما يجدي استخدام الأدوية النفسية في التخفيف من تلك السلوكيات. .jpg) القصور الجنسي: من العلامات المميزة لمتلازمة برادر ولي، حيث يلاحظ : o عدم أكتمال الأعضاء التناسلية الخارجية في الذكور، صغر حجم القضيب، تأخر نزول الخصيتين، وهو ما قد يحتاج للتدخل الجراحي o عادة ما تبدأ التطورات الجنسية في عمر مبكر مثل ظهور شعر العانة، وتتوقف قبل الوصول إلى مرحلة البلوغ. o تأخر نزول الطمث لبعد سن 16أو عدم نزوله لدى الإناث o نقص الهرمونات الجنسية testosterone and estrogen o التدخل العلاجي بالهرمونات ذي نتائج جيدة ولكن هناك ردود فعل سيئة من استخدامه o عادة ما يكون المصابين غير قادرين على الإنجاب، ولكن سجل العديد من الحالات التي حصل بها حمل وانجاب. التشخيص: o الأعراض المرضية، وهناك قواعد معينة للتشخيص من حيث عدد العلامات الرئيسة والعلامات غير الرئيسة في كل مرحلة عمرية، وأخصائي الأمراض الوراثية لديهم هذه القواعد والجداول التابعة لها. o DNA methylation analysis confirms diagnosis o the specific genetic cause and associated recurrence risk FISH and DNA techniques can identify .jpg) .jpg) .jpg) هل يساعد التشخيص المبكر الأطفال المصابين بمتلازمة برادر ويلي ؟ في البداية لا بد من التذكير أن الحالة نتيجة عيوب في الكروموسومات وتلك لا يمكن علاجها أو تغييرها، ولكن التشخيص المبكر يعطي الوالدين الفرصة للتعرف على الحالة والتدرب على مواجهة التحديات المستقبلية، كما البحث عن طرق الدعم والمساعدة، فمثلاً معرفة وجود التأخر في النمو الحركي والفكري سيساعدهم في عدم البحث عن المسببات لهذا التأخر، ومعرفة الطرق للتعامل مع هذا التأخر، كما يمكن التعرف على العائلات ممن لديهم مثل هذه الحالة لمعرفة العقبات التي واجهتهم وطرق التعامل معها ومن أهمها طرق التعامل مع الشراهة في الأكل. ما هو مستقبل الأطفال المصابين بمتلازمة برادر ويلي ؟ مع وجود الكثير من المشاكل الجسمية والسلوكية لدى المصابين بمتلازمة برادر ويلي، ولكن بمساعدة العائلة والمدرسة والمجتمع يمكن لهم الحياة في المجتمع والعمل بصورة أشبه بالطبيعية، ولكن ذلك يحتاج إلى الكثير من الجهد والوقت والتعاون بين جميع المؤسسات ذات العلاقة. في الماضي الأطفال المصابين بمتلازمة برادر ويلي كانوا يتوفون في مرحلة مبكرة، وخاصة في مرحلة المراهقة نتيجة السمنة المفرطة، ولكن مع التحكم في السمنة والتغذية من خلال العلاج النفسي – الغذائي – الدوائي، أمكن لهؤلاء الأطفال من العيش لعمر أطول، والدراسات والأبحاث مستمرة للبحث عن طرق جديدة لمساعدتهم .jpg)
|
|
|
|
|
05-10-2010, 12:11 AM
|
#9 | |
|
متلازمة تيرنرTurner Syndrom النقص في عدد الكروموسوم الجنسي 45XO اعداد الدكتور : عبدالله محمد الصبي - أخصائي طب الأطفال - مدينة الملك عبدالعزيز - الحرس الوطني عرفها الطبيب هنري تيرنر H.H. Turner الأستاذ بجامعة أكلاهوما ونشرها عام 1930، حيث لاحظ على سبعة من مرضاه خصائص متقاربة ، فقد وجد أنهن قصيرات في القامة، لهن جلد زائد على جانبي الرقبة، كما لم تكتمل لديهم الصفات الجنسيّة الأنثوية، وتكرر وصف تلك الحالات من العديد من الأطباء في أنحاء العالم، لذلك سميت الحالة بمتلازمة تيرنرTurner Syndrom ، ولكن لم يتم التعرف على المسبب لوجود الحالة سوى عام 1959م ، عندما تم أكتشاف فحص الكروموسومات، وأن السبب في حدوثها هو فقدان الكروموسوم الجنسي في الأنثى كاملاً أو جزئياً. 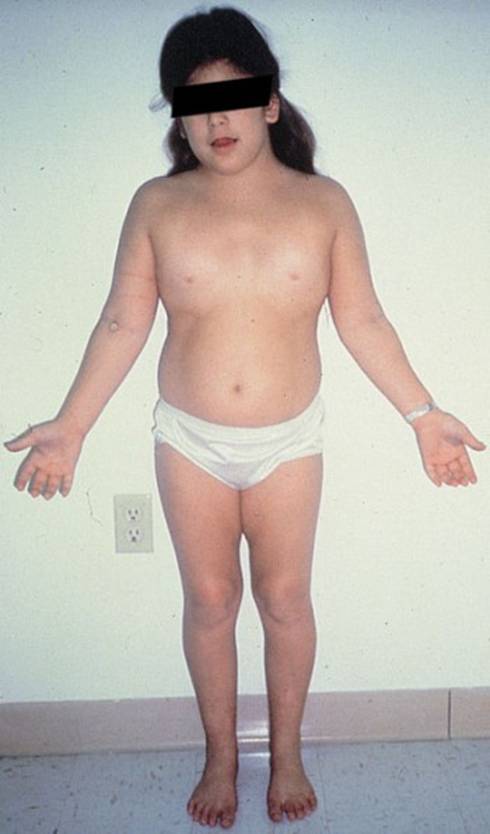 o يصيب هذا المرض الإناث فقط o تحدث حالة واحدة لكل 3000 ولادة لأنثى حيّة o 98 % من الأجنة المصابة يحدث لها أجهاض o ليس له علاقة بسن الأم عند الحمل o ليس له علاقة بالعوامل البيئية الأسباب : o تحدث نتيجة فشل انفصال كروموسوم الجنس أثناء الانقسام meiotic non-disjunction o يكون مجموع كروموسومات الخلية 45 بدلا من 46 o هناك كروموسوم جنسي أنثوي واحد فقط 45XO o قد يكون هناك كروموسومين أنثوي، ولكن أحدهما لا يعمل o في بعض الحالات قد تحمل الأنثى كروموسوم X بجانب بعض مواد من كروموسوم Y o النوع الفسيفسائي- بعض الخلايا طبيعية 46XX - والبعض ناقصة 45XO .jpg) .jpg) الأعراض المصاحبة : o قد لا يكون هناك أي عرض للمرض عند الولادة أولا تكون الأعراض واضحة o انتفاخ اليدين و القدمين يحدث لحوالي 80 % من المواليد المصابة--- وقد يكون هو العلامة الأولى للحالة، تختفي خلال أشهر بعد الولادة. o الأعراض تختلف من شخص لآخر، ولكن تتميز بوجود قصر القامة وغياب المميزات الجنسية o الاعراض الجسمية o مظهر انوثي o عيوب خَلقية في القلب أو الشريان الأورطي في 50% من الحالات o عيوب خلقية في الكليتين في 30-60% من الحالات o اضطراب الغدة الدرقية o ارتفاع ضغط الدم o ضعف في السمع o مشاكل في النظر o تاخر عقلي بسيط o صعوبات التعلم .gif) الأعراض الجسمية : o القامه قصيرة o الرقبة : قصيرة وعريضة، ومن الخلف جلد زائد على جانبي الرقبة، انخفاض حد الشعر في الرقبة من الخلف .jpg) o صغر حجم الفك السفلي، سقف الحلق ضيق o أذن منخفضة وكبيرة ومرتفعة للخارج o العين : زائدة لحمية في الجزء الداخلي، نقص الدموع، ارتخاء الجفن o الصدر عريض وحلمتي الثدي متباعدتين o اتساع وميلان اليد عند المرفق o الأصابع للداخل ، الأصبع الرابع قصير، الأظافر قليلة النمو o الخط المفرد في الكف o بقع جلدية صغيرة بنية اللون .jpg) .jpg) .jpg) المشاكل الجنسية : o غياب للميزات الجنسيه عند البلوغ - الثدي - الشعر - الصوت o لا وجود للدورة الشهرية o الأعضاء التناسلية غير ناضجة o تشوه في الغدد التناسلية o عقم الصعوبات التعليمية: o صعوبة خاصة في الرياضيات non-verbal or spatial learning o قدرة جيدة على الكتابة o التعلم من خلال السمع o قدرة جيدة على التذكر o قدرة جيدة على النطق التشخيص : o العلامات المرضية قد لا تكون واضحة o قد يبدأ الشك في حالة الفشل الدراسي o عدم أنتظام الدورة الشهرية، أو عدم الحمل o التشخيص يكون عن طريق الصورة الكروموسومية Karyotyping o الأشعة الصوتية للبطن : تشوه وضعف المبايض والرحم o الفحص المهبلي - غير طبيعي o ارتفاع الهرمونات Serum luteinizing hormone، Serum follicle stimulating hormone o ارتفاع estriol في الدم والبول هل يمكن التشخيص خلال الحمل وقبل الولادة ؟ نعم - من خلال أجراء بعض الفحوص مثل Chorionic Villus Sampling and Amniocentesis مدى الحياة : o طبيعي في أغلب الحالات o يتأثر بوجود الأمراض المصاحبة (الضغط، القلب ، الكلى) حالات مشابهة : متلازمة نونان Nonan Syndrome، وتلك تنتقل عن طريق الوراثة السائدة Autosomal Domenant، حيث قصر القامة، قصر الرقبة، انخفاض حد الشعر في الرقبة من الخلف، تورم الأطراف .jpg) العلاج : o لا يوجد علاج شافي للحالة، فالعيب في الكروموسومات o الكثير من النساء المصابات بالحالة يعيشون بشكل طبيعي، ويتم اكتشاف الحالة بالمصادفة o إعطاء هرمون النمو Growth hormone في وقت مبكر من الممكن أن يساعد المرأة للوصول إلى طول أقرب للطبيعي. o أعطاء هرمون الأنوثة estrogen بدأ من العاشرة من العمر، من الممكن أن يساعد على ظهور المظاهر الأنثوية كبروز الثديين، وحصول الدورة الشهرية o التلقيح الصناعي in vitro fertilization يمكن ان يساعد على الحمل o الفحص الدوري للسمع والقلب كل سنتين o علاج الحالات المصاحبة ( القلب، ارتفاع ضغط الدم، الغدة الدرقية، الحول ومشاكل السمع والبصر، وغيرها) .jpg) خطط نمو الطول و الوزن للمصابات بمتلازمة تورنر من الولادة و حتى 18 سنة ( المنحنيات الحمراء) (المجالات البيضاء خاصة بالفتيات الطبيعيات من أجل المقارنة ) .jpg) مخطط سرعة النمو عند المصابات بمتلازمة تورنر ( المنحنيات الحمراء) (المجالات البيضاء خاصة بالفتيات الطبيعيات للمقارنة ) .jpg) مخطط كتلة الجسم عند المصابات بمتلازمة تورنر |
|
|
|
|
05-10-2010, 12:13 AM
|
#10 | |
|
متلازمة وولف – هيرشيرون Wolf-Hirschorn syndrome متلازمة الكروموسوم الرابع الناقص Chromosome 4p syndrome الدكتور : عبدالله محمد الصبي – أخصائي طب الأطفال – مدينة الملك عبدالعزيز – الحرس الوطني تم التعرف على الحالة عندما قام الدكتور كيرت هيرشينون Kurt Hirschhorn وهيربيرت كوبر Herbert Cooper عام 1961 بنشر دراستهم ، حيث وصفوا حالة طفل لديه فشل في الالتحام في منطقة منتصف الجسم مع وجود عيب في تركيب كروموسوم الخلية المتمثل في نقص الذراع القصير للكروموسوم رقم 4 deletion ، وفي عام 1965 نشر بحث هيرشينون - وولف Wolf-Hirschhorn الذي سلط الضوء على هذه المتلازمة للعاملين في القطاع الطبي، ومن ثم تمت تسمية الحالة بإسميهما. 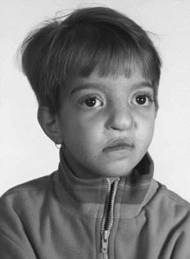 هذه الحالة تحدث نتيجة وجود فشل في إلتحام أجزاء الجسم المركزية ( الخط الوسطي الطولي للجسم )، وتتميز هذه الحالة بصغر حجم الرأس، التخلف الفكري، الصرع، العلامات المميزة للوجه ، كما علامات أخرى متنوعة. .jpg) o نسبة حدوثها في أمريكا هو حالة لكل 50,000 ولادة o تصيب كل الأجناس وكل البلدان o تصيب الإناث ضعف الذكور o عادة ما يتم الشك فيها عند الولادة لوجود الأعراض الظاهرة o نسبة عالية يحدث لها اجهاض o نسبة الوفيات 34% في السنتين الأوليتين o أسباب الوفاة : العيوب القلبية، الصرع، الالتهابات، الاصابات الرئوية o في عدم وجود عيوب خلقية كبيرة ، فليس هناك ما يؤدي للوفاة، ويستطيع المولود العيش حياة طويلة الأسباب : o السبب غير معروف – ليس للوراثة دور في حدوثه o ليس هناك تأثير لعمر الوالدين على حدوث هذه المتلازمة o العدد الكلي للكروموسومات طبيعي ، فليس هناك نقص في العدد الكلي o تحدث نتيجة نقص - حذف deletion للذراع القصير short arm من الكروموسوم في المجموعة 4 o الجزء المؤثر الرئيسي هو نقص الجزء 4p16.3 o السبب في 87% من الحالات هو نقص – ذكوري الأصل o السبب في 13% من الحالات - من المنوع المتحول الأصل a reciprocal translocation .jpg) الأعراض خلال الحمل : o ضعف نمو الجنين o ضعف حركة الجنين o صغر حجم المشيمة الأعراض المرضية : o ضعف عام للنمو o قصر القامة o صغر حجم الرأس o صعوبات في التغذية والتنفس .jpg) العلامات الجسمية : o الأصابع طويلة ونحيفة، زيادة عدد الخطوط في الأصابع، وجود أصبع زائد – خاصة الإبهام o وجود الخط المنفرد في الكف 25% o تباعد حلمتي الصدر o عيوب خلقية في عظام القفص الصدري وعظام العمود الفقري o ضعف نمو العظام .jpg) الرأس : o الرأس : صغر حجم الرأس، كبر حجم الجبهة، الوجه المميز Greek warrior helmet، o عيوب في فروة الرأس o الأنف المفلطح o العينيين : جحوض العينين، صغر حجم العين، وجود ثنيات لحمية في العينين، ميلان العينين إلى الأسفل، الحول، نقص في القزحية coloboma، الماء الأبيض – الساد o صغر حجم الشفة العليا o الشفة الارنبية وشق الحنك o صغر حجم الفك السفلي o الأذن كبيرة، مشوهة، وهابطة عن مكانها o نقص السمع العصبي o العيوب القلبية : فتحة بين البطينين، فتحة بين الأذينين، عيوب مركبة في القلب o عيوب خلقية في الرئتين o عيوب خلقية في الجهاز الهضمي o عيوب خلقية في الجهاز البولي والتناسلي .jpg) الجهاز العصبي والحركي : o صغر حجم الرأس o عيوب خلقية في الدماغ o الاستسقاء الدماغي o تخلف فكري شديد o الصرع والتشنج o أرتخاء عام للعضلات العلامات التطورية والنفسية : o ضعف التطور الحركي والفكري o صعوبة الحركة ، الحركات غير المتوازنة ataxic gait o الصرع 50% o تأخر النطق – عدم الكلام o النغنغة أو الأصوات غير ذات معنى o نقص في التواصل ، ويكون التواصل عن طريق الحركات والتعبيرات o الحركات غير الطبيعية لليدين مثل وضع اليدين أمام الوجه، تحريك اليدين بحركة الغسيل، الضرب على الصدر، تحريك الرأس المستمر o يستطيع تناول الأكل بنفسه o يساعد على ارتداء ملابسه حالات مشابهة : الصفات الظاهرة للحالة قد تتشابهة مع حالات أخرى، لذى فإن التشخيص يعتمد على التحليل الكروموسومي، وهذه الحالات المشابهة هي : o متلازمة باتو Patau Syndrome - تثلث الكرموسوم رقم 13 o متلازمة ادوارد Edwards syndrome - تثلث الكرموسوم رقم 18 o متلازمة مواء القطط Cri-du-chat Syndrome o متلازمة سميث – ليميلي – أوبيتز Smith-Lemli-Opitz Syndrome .jpg) التشخيص : يعتمد التشخيص على أكتشاف العطب في الكروموسوم وليس الأعراض الظاهرة، ويعتمد التشخيص على مجموعة من الاختبارات منها : o الصورة الكروموسومية العادية غالباً ما تكون طبيعية ، ولا يتم اكتشاف الحالة من خلالها o Conventional cytogenetic studies : وهي الطريقة المثلى لإكتشاف النقص الحاصل في ذراع الكروموسوم 4p، ولكن يعيبه إحتمالية عدم اكتشاف النقص في الذراع القصير 4p o High-resolution cytogenetic studies : حيث يمكن اكتشاف النقص في أجزاء الكروموسوم 4p16.3 o Fluorescence in situ hybridization – FISH - .jpg) .jpg) أختبارات أخرى : هناك العديد من الاختبارات والتحاليل يمكن أجرائها ليس بغرض تشخيص الحالة ولكن تشخيص الأعراض المصاحبة ، مثل : o أختبارات المناعة o أشعة صوتية للقلب Echo-Cardiography o أشعة صوتية للجهاز الهضمي والبولي Ultrasound o أشعة مقطعية ، الرنين المغناطيسي للدماغ CT-scan, MRI o تخطيط الدماغ EEG في حالات الصرع o أشعة ملونة للجهاز الهضمي – مثل الأشعة الخاصة بالبلع o قياس السمع والنضر العلاج : لا يوجد علاج للحالة، فالمشكلة عيب خلقي في الكروموسوم يؤثر على جميع خلايا الجسم، وتلك لا يمكن علاجها أو تغييرها، ولكن يمكن علاج الأعراض المصاحبة للحالة من خلال الفريق الطبي ، مثل : o علاج الصرع بالأدوية o العيوب الخلقية في القلب o العيوب الخلقية للعين o النطق والتخاطب والسمع o العلاج الطبيعي والعلاج الوظيفي الأستشارة الوراثية : o نسبة تكرار الحالة ضعيف جداً o تزيد نسبة تكرار الحالة اذا كان أحد الوالدين يحمل عيباً جينيا – وراثياً – مثل النوع المتحول المتوازن translocation carrier. هل يمكن التشخيص خلال الحمل وقبل الولادة ؟ نعم، يمكن التشخيص خلال الحمل وقبل الولادة، ولكن يجب الشك في وجود الحالة من خلال حمل سابق مثلاً، مع العلم أن نسبة التكرار ضعيفة، ومن طرق التشخيص قبل الولادة : o الأشعة الصوتية للجنين Ultrasound : قد يظهر الأعراض المرضية الظاهرية للحالة، مثل ضعف النمو للجنين، صغر حجم الرأس، شق الحنك، فتق الحجاب الحاجز، ولكن تلك العلامات غير تشخيصية وتحتاج للتحليل الكرموسومي الخاص. o تحليل السائل الأمنيوسي – ماء الجنين- Amniocentesis: خلال الأسبوع 14-16 من الحمل. o تحليل عينة المشيمة Chorionic villus sampling: خلال الأسبوع 10-13 من الحمل o تحليل عينة من دم الجنين Percutaneous umbilical blood sampling .jpg) .jpg)
|
|
|
|
|
05-10-2010, 12:14 AM
|
#11 | |
|
متلازمة إدوارد اعداد الدكتور : عبدالله محمد الصبي – أخصائي طب الأطفال – مدينة الملك عبدالعزيز – الحرس الوطنيEdward syndrome ( التلث الصبغي رقم 18 ) تم التعرف على هذه الحالة عندما قام الدكتوران ادوارد و سميث Edwards et al and Smith et al عام 1960بنشر بحث عن حالات متشابهة في الأعراض المرضية مثل صغر الحجم عند الولادة ، التخلف الفكري والحركي، صغر الفم و الفك السفلي، عيوب صيوان الأذن، عيوب في الكفين والاصابع، عيوب خلقية في القلب، عدم هبوط الخصيتين، وغيرها، وأن السبب في ذلك هو وجود كروموسوم زائد في المجموعة رقم 18 من الكروموسومات. 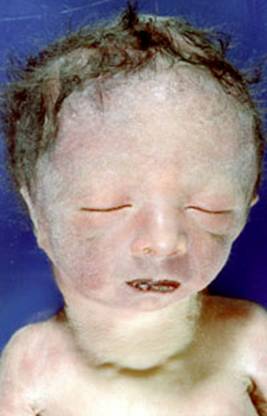 o ناتجة عن زيادة في العدد الإجمالي للكروموسومات – هناك47 كروموسوم - تكون الزيادة في المجموعة رقم 18، سواء كانت تلك الزيادة كاملة أو جزئية o نسبة حدوثها حالة لكل 20.000-40.000 ولادة طفل حي o نسبة الإناث المصابات ثلاث أضعاف عدد الذكور o جميع الاجناس وجميع الدول o أغلب الحالات تنتهي بالاجهاض o هذه الزيادة في جميع خلايا الجسم، ومن ثم تؤثر في جميع أجهزة الجسم o الأعراض المرضية تختلف درجتها وحدتها من مصاب لآخر، فقد لوحظ أن الحالات من النوع المتحول translocations أو النوع الفسيفسائي mosaic أقل حدة وأعراض، ولذلك فمن الممكن لهم أن يعيشوا لمدة أطول الأسباب : o السبب في حدوث المتلازمة غير معروف o أغلب الحالات 95% تظهر بالصورة الكاملة nondisjunction o النوع الفسيفسائي Mosaicism يحدث في 3% من الحالات o النوع المتحول translocations قليل جداً o يلعب عمر الأم دوراً في زيادة حدوث المتلازمة o في 90% من الحالات وجد أن الزيادة من خلايا الأم، والمشكلة تحدث في الانقسام الثاني للخلية meiosis II ضعف الانقسام الأولي meiosis I، وهو عكس ما يحدث في الانواع الأخرى من التثلث الصبغي o يحدث عدم الانقسام nondisjunction في الذكور نتيجة في مرحلة بعد الالتحام postzygotic mitotic o النوع الفسيفسائي mosaic يحدث غالبا نتيجة مرحلة بعد الالتحام postzygotic mitotic o وجد أن الجزء المؤثر من الكروموسوم المؤدي للأعراض المرضية هو 18q11-q12. الأعراض : o عند الحمل – زيادة حجم الرحم الناتج من زيادة كمية السائل الامنوسي o صغر في الحجم و البنية عند الولادة (الوزن و الطول و محيط الرأس) .jpg) الرأس والرقبة : o صغر الرأس o بروز مؤخرة الرأس o صغر فتحة العينين o وجود ثنية جلدية للركن الخارجي لجفن العين Espiscanthal folds o فقد جزء من قزحية العين Coloboma of iris o انخفاض مستوى الأذنين عن مستوى العينين مع قله في طوية الأذن الخارجية o صغر الفم و الفك السفلي micrognathia o زيادة احتمال أن تحدث الشفة الارنبية و الحلق المشقوق o ثنيات جلدية زّائد في مؤخرة الرقبة .jpg) .jpg) .jpg) اليدين والقدمين : o تراكب أصابع اليدين بشكل مميز(انطباق السبابة على الإصبع الوسطى و فقها الإبهام) o الالتصاقات بين الأصابع o صغر الأظافر o صغر إبهام اليد و الرجل مع احتمال غيابها o قصر أو تقوس إبهام القدم إلى الخلف Dorsiflexed o غياب الثنية البعيدة في الإصبع الصغيرة(الخنصر)و قد تؤدي إلى انحناء الإصبع إلى الداخل o تقوس باطن القدمين إلى الخارج o غياب احد عظمات الساعد في اليدين في حوالي 10% من المصابين o قصر عظم القص (العظم الذي يربط بين ضلوع الصدر ) o تيبس في المفاصل(Joint contractures) .jpg) .jpg) .jpg) .jpg) عيوب خلقيّة في القلب : o تحدث في 90% من الحالات o فتحة بين البطينين VSD -ventricular septal defect o فتحة بين الأذنين ASD -atrial septal defect o فتحه جنينية بين الأبهر والشريان الرئوي patent ductus arteriosus- PDA عيوب خلقية في الجهاز البولي والتناسلي : o عدم نزول الخصيتين o عيوب في الشكل مثل Horseshoe kidney o الارتجاع البولي Hydronephrosis o الكلى المتكيسة Polycystic kidney عيوب في أجهزة الجسم الأخرى : o ضعف السمع o عيوب خلقيّة في الرئتين و الحجاب الحاجز o الظهر المشقوق 6% o الفتاق و / أو انفصال عضلات جدار البطن المشاكل الصحية المصاحبة : o ضعف النمو و قصر القامة o صعوبات التغذية و الترجيع المعدي إلى المريء o الارتخاء العام 100% وخاصة في مرحلة الطفولة المبكرة o التخلف الفكري 100%( من النوع الشديد في جميع المصابين تقريباً ) o التأخر الحركي 100% o التشنجات و حالات الصرع 30% o المشاكل المتعلقة بعيوب القلب o توقف التنفس المتكرر(Central apnea ) o المشكل المتعلقة بالكلى و ارتفاع ضغط الدم o انحناء و تقوس الظهر التشخيص: o يتم تشخيص المرض إكلينيكيا عن طريق الأعراض و العلامات الخارجية للطفل o يتم التأكد من التشخيص عن طريق إجراء تحليل للكروموسومات و ذلك عن طريق زراعة خلايا الدم .jpg) الأنواع والأختلافات في الصورة الكروموسومية : التثلث الصبغي في المجموعة رقم 18 ليست صورة واحدة، ولكن العديد من الصور، وهو ما يحتاج لمتخصصين، وللتوضيح سندرج هذه الأنواع بدون الدخول في التفاصيل، وهي : NORMAL .gif) 18P .gif) 18q .gif) Ring Form .gif) Tetrasomy 18p .gif) Trisomy 18 .gif) تشخيص الأعراض المصاحبة : o الأشعة الصوتية للقلب لمعرفة وجود عيوب قلبية o أشعة ملونة للجهاز الهضمي لمعرفة وجود عيوب خلقية o أشعة صوتية للجهاز البولي التناسلي o أشعة للعظام لمعرفة وجود عيوب خلقية التشخيص خلال الحمل : o عادة لا يتم فحص الحمل التالي في حالات التثلث العادي ، ولكن خلال الحمل هناك علامات دالة على وجود المتلازمة مثل : زيادة حجم السائل الأمنوسي – الجنيني polyhydramnios وهو ناتج عن ضعف القدرة على المص والبلع، قلة السائل الجنيني وعادة ما يحدث نتيجة هبوط عمل الكلى، صغر حجم المشيمة، ضعف النمو الجنيني Intrauterine growth retardation، ضعف حركة الجنين، وهو ما قد يستدعي أجراء الفحوصات الأخرى والشك في وجود الحالة. o في حالات النوع المتحول translocational trisomy ، يحتاج الحمل الجديد إلى متابعة، ولك لأرتفاع أحتمالية تكرار الحالة o أختبار السائل الأمنوسي amniocentesis خلال الأسبوع 14-16 للحمل o عينة من الخلايا الجنينية Chorionic villus sampling خلال 10-13 للحمل العلاج : o لا يوجد علاج شافي للمرض ولكن للأعراض o نظراً لحدوث الوفاة مبكرا فان الأطباء لا يقومون بإجراء العمليات الجراحية خاصة المتعلقة بالقلب o معظم الأطفال يتوفون نتيجة لتكرار توقف التنفس من مركز التنفس في المخ(Central apnea) o الأطفال الذين يعيشون ، فقد يراجع الأطباء حالة الطفل و على ضوئها يقومون بإجراء ما يلزم كعملية وضع أنبوب تغذية في المعدة أو إجراء عملية للقلب أو إعطاء أدوية للتشنجات، علما أن هذه الإجراءات لا تقدم كثير و لا تأخر في وضعهم الصحي بشكل عام o الأطباء في كثير من الأحيان لا يقومون بإجراء أي إنعاش للقلب أو إجراء تنفس صناعي إذا لا قدر الله و توقف قلب الطفل أو تنفسه o العائلة تحتاج للدعم النفسي، الدعم الاجتماعي، والدعم المادي o الاستشارة الوراثية مهمة لتوضيح الأمر للوالدين عن الأسباب، التكرار، وكيفية التعامل مع الطفل o قد يحتاج الطفل المصاب إلى المساعدة من العديد من المتخصصين، وذلك يقرره الطبيب المعالج احتمالات تكرار المرض : o يمكن أن تتكرر الحالة بنسبة 1% فقط، وهي نسبة صغيرة o أغلب الحالات تحدث نتيجة طفرة جينية o يمكن أن نقول لا خوف من الحمل التالي بإذن الله، ولا يحتاج الأمر لأجراء أي تحاليل أو أشعات خلال الحمل القادم o يمكن اجراء بعض التحاليل للأمهات الذين تمت ولادة طفل مصاب خلال الأسبوع العاشر من الحمل للتأكد من سلامة الجنين، و لو كان الجنين مصاب فليس هناك حلا علاجيا إلا إجهاض الحمل إذا كان ذلك جائزا شرعا. o تزيد نسبة تكرار الحالة عندما يكون أحد الوالدين يحمل عيباً في الكروموسومات بدون أعراض وهو ما يسمى a balanced carrier، وتلك الحالات يقررها الطبيب المعالج مآل الحالة – أحتمالية الوفاة : o 95% من المصابين بهذا المرض يتوفوا خلال الحمل أو بعد بضعة أيام من الولادة o أغلبية المولودين بهذه الحالة يتوفون خلال الشهر الأول من العمر o 5-10% فقط من المصابين يعيشون إلى نهاية السنة الأولى من العمر و أكثرهن من الإناث o سبب الوفاة هو وجود العيوب الخلقية في القلب والكلى وغيرها o الحالات التي تعيش لمدة طويلة عادة ما يكون لديها تخلف حركي وفكري شديد بالاضافة لصعوبات التغذية والمشاكل الجسمية المتعددة |
|
|
|
|
05-10-2010, 12:15 AM
|
#12 | |
|
متلازمة اهلرز – دانلوس الدكتور : عبدالله محمد الصبي - أخصائي طب الأطفال - مدينة الملك عبدالعزيز - الحرس الوطنيEHLERS DANLOS SYNDROME هي اضطراب وراثي في النسيج الضام connective tissue disorders ، خيث قام بوصفه والكتابة عنه طبيب الجلدية الهولندى أدوارد أهلر Edward Ehlers عام 1901، ثم قام العالم الفرنسي هنري دانلوس Henri-Alexandre Danlos بشرح العيوب الكيمائية للمرض عام 1908 ، لذلك سميت الحالة بأسم هذين العالمين. تتميز هذه الحالة بما يلي : o فرط حركة المفاصل o فرط مرونة الجلد o هشاشة منتشرة بأنسجة الجسم الوراثة : o تورث عادة كسمة صبغية جسمية سائدة o غالباً ما تنتج عن طفرات جينية مختلفة – أي لا يوجد حالات مماثلة في العائلة o وصفت لهذه المتلازمة 10 أنواع مختلفة o يوجد عوز في أنزيم ليزيل هيدروكسيلاز o تؤثر على مجموعة مختلفة من الكولاجينات o يؤثر على كل الأعراق o نسبة الأصابة حالة لكل 10.000 فرد الأعراض : على الرغم من أنه قد تحدث مضاعفات متعددة ومختلفة فإن معدل العمر يكون طبيعياً، ومعدل الذكاء طبيعي، انتشار المضاعفات يكون عالياً في عدد قليل من الأسر فقط، وتختلف الأعراض بشكل واسع حسب الطفرة الجينية الخاصة والنمط الناتج من متلازمة اهلرز-دانلوس، وعادة ما تكون متشابهة في العائلة الواحدة، لذى يلاحظ أنماط ودرجات متنوعة من الحالات: o تمطط الجلد لعدة سنتمترات ولكنه يعود إلى وضعه الطبيعي بعد تركه 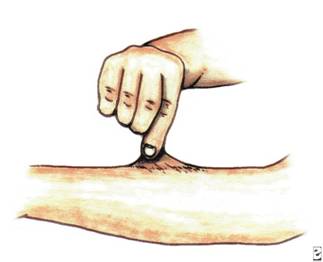 .jpg) o فرط الحركة في المفاصل أصبحنا نلاحظ وجود الأشخاص المصابين بهذه المتلازمة في استعراضات السيرك - النساء المرنات - رجل الهند المطاطي .jpg) .jpg) .jpg) .jpg) .jpg) .jpg) .jpg) .jpg) o ميل للنزف نتيجة للجروح والكدمات - ولكن النزف عادة م ا يكون بسيطاً ، ويكون شديداً في حالات قليلة o قد يسبب الرض الطفيف جروح فاغرة واسعة الفرجة لكنها قليلة النزف، وقد يكون اغلاق الجرح صعباً، نظراً لأن القطب الجراحية تميل لتمزيق النسيج الهش o تنشأ مضاعفات جراحية بسبب هشاشة النسيج العميق o كثيراً ما تتشكل ناميات لحمية ( الأورام الرخوانية الكاذبة ) على رؤوس الندبات أو في نقاط الضغط . o قد نجد كريات متكلسة تحت الجلد أو يثبت وجودها شعاعيا مضاعفات محتملة : o القدم المسطحة في 90% .jpg) o الورك الولادي في 1% o الحدب الجنفي الشوكي 25% o تشوه صدري في 20% o الفتق المعدي المعوي .jpg) مضاعفات محتملة للأم – الحمل والولادة هذه المضاعفات تحدث نتيجة هشاشة أنسجة الأم المصابة بالمرض، لذى ينصح بالمتابعة للحامل في المراكز المتخصصة للولادة. o ولادة الخدج o العملية القيصرية o أخطار خزع الفرج o النزف قبل وحول وبعد الولادة o التمزق الباكر لجيب المياه o قد تسبب قابلية تمدد النسج عند الأم إلى إلى. حالات مشابهة : o متلازمة مارفان Marfan Syndrome مرض وراثي ينتقل من جيل لأخر عن طريق ما يسمى بالوراثة السائدة autosomal dominant trait ، تحدث الأعراض نتيجة لخلل في مورث- جين- يسمى بمورث الفيبريلين رقم واحد(Fibrillin 1) الموجود على الذراع الطويلة لكروموسوم chromosome 15q21.1.، و تدخل هذه المادة في تركيب النسيج الضام ، ومن ثم في تركيبة العديد من أعضاء الجسم كالأوعية الدموية، صمامات القلب،العظام، الجلد، والعين . تختلف الأعراض المرضية بين شخص وآخر، كما تختلف بين أفراد العائلة الواحدة المصابين بالحالة، فقد نجد لدى البعض عرض أو أثنين، وقد تكون شديدة لدى الآخرين، ومن أهم الأعراض : طول القامة والأطراف، الشكل المميز للرأس والوجه، التغيرات في العمود الفقري والصدر، مشاكل العين، مشاكل في الجهاز الدوري والقلب، كثرة حدوث الفتوق الاربية والفخذية، خلوعٍ المفصل المتكرر، تخلف عقلي بسيط في بعض الحالات، صعوبة بالتعلم في بعض الحالات غير المصابة بتخلف عقلي o متلازمة وليامز Williams Syndrome طفرة جينية - حالات قليلة تكون وراثية ، نقص أو حذف جزء من الكرموسوم - الصبغي - السابع q11، من أعراضه : ملامح الوجه مميزة، التخلف الفكري البسيط الى المتوسط، صعوبة النطق والكلام، زيادة نسبة الكالسيوم في الدم، مشاكل القلب والجهاز الدوري، مشاكل التغذية، فتق السرة وفتق اربي، مشاكل الجهاز الحركي - العظام والعضلات التشخيص : يتم التشخيص من خلال : o الأعراض o أستبعاد الحالات المشابهة مثل متلازمة مارفان o عينة الجلد – histopathologic analysis عادة ما تكون غير تشخيصية o بعض الأنواع يتم تشخيصها بالتحليل الكيميائي للنسيج الضام العلاج : o لا يوجد معالجة خاصة للمرض o يجب الإقلال من الرض - الأصابات o قد يكون من المفيد لبس اللباس الواقي والحشوات في الملابس o يجب إجراء ارقاء دقيق جداً عند إجراء العمليات الجراحية، ويجب خياطة الجروح بحذر كما يجب أن نتجنب توتير النسج. o الإشراف الطبي خلال الحمل والولادة يكون إجبارياً o يجب إعطاء الاستشارة الوراثية |
|
|
|
|
05-10-2010, 12:16 AM
|
#13 | |
|
Aase syndrome أدهم أحمدAase-Smith syndrome التعريف : هو مرض وراثي نادر و من اهم خصائصه انه يكون مصحوب بالانيميا (فقر الدم) مع عدم انتظام في شكل العظام و المفاصل. " الاسباب و عوامل الخطر: يرجع الكثير من العلماء السبب في خلل في احد الكرموسومات الجسدية و لا تزال القاعده الاساسية للاصابة بهذا المرض مجهولة (توجد في بعض العائلات النادرة) و بالنسبة لفقر الدم الدم يرجع لعدم نضوج نخاع العظام حيث مكان تصنع خلايا الدم بأنواعها المختلفة " الأعراض: 1.النمو البطيء للجسم 2.شحوب لون الوجه و الجلد بشكل عام و ذلك بسسب فقر الدم 3.تأخيرو بطء في اغلاق الفجوات في الرأس عند الولادة fontanel's 4.قصر و ضيق الكتفين 5.وجود 3 مفاصل في اصبع الابهام 6.عدم قدرة فرد الاصابع بسبب عدم انتظام المفاصل 7.عدم انسداد سقف الفم بحيب يكون مفتوح التجويف الانفي على الفم 8.عدم انتظام غضاريف الاذنين 9.سقوط جفوف العينين " الفحوصات التي يتم اجرائها في هذه الحالة هي : 1.فحص دم شامل : نجد فقر دم و انخفاض في مستوى خلايا الدم البيضاء الدفاعية 2.الايكو : و تكون النتيجة وجود عيوب خلقية في القلب و خصوصا في الحاجز الواقع بين الاذيننين 3.الاشعة السينية : و تظهر الخلل الواقع في العظام و المفاصل و عدم انتظام جزيئات العظام 4.سحب عينة من نخاع العظم : يظهر بطء نمو النخاع " العلاج: 1.نقل الدم للطفل لعلاج فقر الدم و ذلك في العام الاول 2.اعطاء بريدنزون و هو من أحد أنواع الكوتيزون .. العلاج السحري 3.ولكن ممنوع اعطائه في مرحلة الطفولة و ذلك لتأثيره المباشر على طبيعة النمو الجسم و خصوصا الدماغ 4.والعلاج النهائي في حالة فشل العلاجات السابقة هو زراعة نخاع العظم " توقعات مستقبلية بالنسبة لهذا المرض: 1.هو التخلص من فقر الدم و علاجه " المشكلات التي ممكن أن تصيب المريض : 1.استمرار فقر الدم و ظهور الاعراض العامة لها مثل التعب و الارهاق السريع عند بدل اي مجهود و الضعف العام و مشاكل الجهاز التنفسي 2.انخفاض مستوى خلايا الدم البيضاء و زيادة احتمال الاصابة بامراض مختلفة 3.فشل في عمل عضلة القلب و حدوث الكثير من المضعفات اثر ذلك 4غالبا ما يموت الجنين قبل الولادة أو بعد الولادة بفترة زمنية قصيرة. " منع حدوث هذا المرض: 1.لا يمكن منع هذا المرض الا بواسطة: 2.عدم الزواج الاقارب 3.عدم الزواج من العائلات التي لها تاريخ بحدوث مثل هذا المرض مما يلزم الشاب أو الفتاه المقبل على الزواج تقصى حدوث هذا المرض في تلك العائلات سابقا 4.التوكل على الله في كل شي و عند الاقبال على الخطوبة و الدعاء أن يرزقك الله أطفال أصحاء و ذرية صالحة تنفع الاسلام و المسلمين لا أن تكون عاله عليهم. |
|
|
|
|
05-10-2010, 12:18 AM
|
#14 | |
|
متلازمة باتو Patau Syndrome الدكتور عبدالله محمد الصبي - أخصائي طب الأطفال - مدينة الملك عبدالعزيز - الحرس الوطني تثلث الصبغي 13 حالة متلازمة باتو قديمه من حيث التشخيص الإكلينيكي، ولكن لم يتم التعرف على المسبب وهو وجود الكرسوم الزائد في رقم 13 سوى عام 1960، وتعتبر من أقل حالات التثلث الصبغي حدوثاً وذلك لارتفاع نسبة الإجهاض لهذه الحالة، كما أن نسبة الوفيات المبكرة عالية جداً، ويعتبر التخلف الفكري الشديد سمه ثابتة لتلك الحالات. 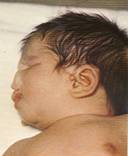 .jpg) o ثلاثي الكروموسوم رقم 13 o لديه ثلاث نسخ من كروموسوم 13 o نسبة حدوثها 1 من كل 12000 مولود حي o تحدث لكلا الجنسين o السبب غير معروف o يؤثر عمر الأم في حدوث الحالة o نسبة حدوثها عالية جداً، ولكن اغلبها تنتهي بحدوث الإجهاض o نادراً ما يعيش الطفل أكثر من ستة أشهر من العمر .jpg) .jpg) الأعراض: الرأس والرقبة: o شفة مشقوقة مع حنك مشقوق o صوان الأذن منخفض وأذان مشوهه o صغر الفك السفلي o صغر حجم الرأس، مع عيوب في فروة الرأس o صغر حجم العين o تخلف عقلي شديد o نقص في السمع أو صمم .jpg) اليدين والقدمين: o أصابع الأقدام والأيدي الإضافية o القدم المهزة Rocker-bottom feet .jpg) .jpg) .jpg) .jpg) أجهزة الجسم الأخرى: o عيوب خلقية في الجهاز التنفسي والجهاز الهضمي o عيوب خلقية في القلب o الفتق o عادة يحدث الموت في الطفولة المبكرة التشخيص: o عن طريق اختبار الكروموسومات بعد الولادة مباشرة o زيادة في عدد الكروموسومات رقم 13 - nondisjunction during maternal meiosis o ممكن الكشف أثناء الحمل من خلال أخذ عينة من ماء الرحم .jpg) .jpg) FISH الحالات المشابهة - التشخيص الفارق: o متلازمة ادوارد - ثلاثي الكروموسوم رقم 18 Edwards syndrome o الزيادة الجزئية في الكروموسوم رقم 13 - Partial duplication of 13q مدى الحياة : o 82 % من الأطفال الرضع يموتون في شهورهم الأولى o 5-10 % يبقون على قيد الحياة إلى سنة واحدة o نادراً ما يعيش لسن الرشد .jpg) العلاج: o لا يوجد علاج للحالة - لوجود العيوب في الكروموسومات o التشوهات الجسمية يمكن أن تعالج بالجراحة o العلاج الطبيعي، والعلاج الوظيفي، علاج النطق .jpg)
|
|
|
|
|
05-10-2010, 12:19 AM
|
#15 | |
|
متلازمة دي جورج الدكتور عبدالله الصبيDiGeorge Syndrome أخصائي طب الأطفال ذوي الاحتياجات الخاصة قام الدكتور انجيلو دي جورج عام 1960 Angelo DiGeorge بوصف مجموعة من الحالات تتشابه وتتزامن فيها بعض الأعراض المرضية، ولذلك سميت هذه المتلازمة باسمه، وفي عام 1982م أثبت العالم كيلي وزملاؤه Kelly العلاقة القوية بين التشوهات في الكرموسوم 22 ومتلازمة دي جورج، وتعتبر هذه المتلازمة من عيوب تطور الخيشوم الثالث والخيشوم الرابع، كما تسمى velocardiofacial syndrome، وحديثاً تم إضافة حالات لهذه الحالة مثل CHARGE، Opitz-GBBB syndrome 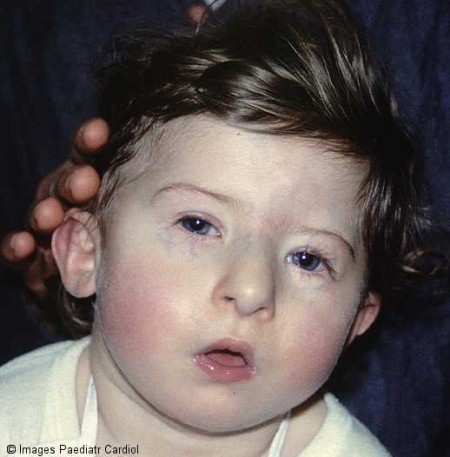 وتشمل متلازمة دي جورج المشاكل التالية: o ضمور الغدد جار الدرقية Para Thyroid Gland o ضمور الغدة السعترية -الغدة التيموسية Thymus Gland o عيوب خلقية في القلب o وجود الشفة الأرنبية Cleft Lip وتشوهات كلوية ورئوية o الصعوبات النفسية والاجتماعية نسبة الانتشار: يعتقد أن نسبة انتشار الحالة تصل لحالة لكل ثلاثة آلاف ولادة، ولكن الأعراض لا تكون واضحة في الكثير من الحالات، ويصيب الذكور والإناث بنفس النسبة يولد طفل مصاب بهذا المرض لكل 68 طفل مصاب بعيوب خلقية في القلب يولد طفل مصاب بهذا المرض لكل 20 طفل لدية شق في سقف الحلق الأسباب: في عام 1982م أثبت العالم كيلي وزملاؤه العلاقة القوية بين التشوهات في الكرموسوم 22 ومتلازمة دي جورج باستخدام تقنية تهجين الفلوريسنت Fluorescent In Situ Hybridization إن الحالة ناتجة عن نقص جزئي في الذراع الطويلة لكروموسوم 22 ( 22q11) بنسبة 95% من الحالات أو نقص في الذراع الطويلة لكروموسوم 10 في 5% من الحالات السبب الحقيقي وراء هذا النقص غير معروفة، ولكنه معروف انه حدث عند انقسام الخلية عند خلق البويضة أو الحيوان المنوي- أي قبل تلقيح. إن معظم حالات متلازمة دي جورج ناتجة عن نقص "طفرة" في كروموسوم 22 ، و هذا يعني أن الطفل فقط هو الذي لديه نقص بينما كروموسومات الأبوين سليمة. في حوالي 10% من الحالات يكون هذا النقص موجود في جميع خلايا الأب أو الأم مع عدم ظهور أعراض واضحة في بعض الأحيان. و إذا كان الحال كذلك فإن هناك احتمال إصابة طفل آخر بمتلازمة دي جورج ، وهذا ما يعرف بالوراثة السائدة، لذلك يتم إجراء تحليل الكروموسومات للوالدين يمكن حدوث الحالة نتيجة التعرض لبعض المواد أثناء فترة الحمل كالكحول ومشتقات فيتامين ( أ ) والحمل السكري .jpg) .jpg) الأعراض: هناك تفاوت واختلاف كبير بين المصابين بمرض دي جورج من ناحية شدة الإصابة من طفل لآخر، كما الآثار المترتبة عليها أو التي تنتج في المستقبل، وعلى العموم يمكن وجود الأعراض التالية بدرجات متفاوتة: أولاً: الشكل العام: الشكل العام غير تشخيصي، ولا يتواجد في جميع الحالات، ومنها: o صغر حجم الرأس، بروز الجبهة hypertelorism o صغر حجم الحنك micrognathia o صغر حجم الفم fish-mouth o ميلان العيون للداخل والأسفل antimongoloid slant o عيوب في صيوان الأذن وصغره o شق الحنك والشفة الأرنبية .jpg) .gif) .jpg) ثانياُ: ضمور الغدد جار الدرقية Para Thyroid Gland نقص هرمون الغدة الجنب درقية Parathyroid hormone الذي يسبب نقص مستوى الكالسيوم بالدم وارتفاع الفسفور، ويؤدي لحالات التشنج ثالثاً: ضمور الغدة السعترية -الغدة التيموسية Thymus Gland ينتج عن هذا نقص في جهاز المناعة الخلوية o وهو أحد أسباب الوفاة الفجائية عند نقل الدم في بداية عملية القلب الجراحية في حالة إغفال هذا النقص المناعي o الطفل معرض للإصابة بالالتهابات بشكل أكثر من اقرأنه الأسوياء ، ويبدو كثير الزكام و الرشح والتهابات الإذنين والنزلات المعوية والإسهال . o قد يتعرضون لموت مفاجئ عند الإصابة بالتهابات شديدة .jpg) رابعاً: عيوب خلقية في القلب: عيوب خلقية في القلب، ضيق أو ضمور أو انقطاع في الشريان الأورطي، الجذع الشرياني أو إي نوع من العيوب القلبية، وقد تكون هي السبب في البحث عن الحالة وتشخيصها 1. رباعية فالوت Tetralogy of Fallout (17%) 2. فتحة بين البطينين ventricular septal defect - 14% 3. انقطاع في الشريان الأورطي interrupted aortic arch - 14% 4. ضمور الشريان الرئوي pulmonary atresia 10% .jpg) .jpg) خامساً: مشاكل أخرى: o التأخر في نمو الطفل المصاب o تشوهات كلوية o تشوهات رئوية o صعوبات التعلم o الصعوبات النفسية .jpg) التشخيص يبدأ الطبيب بالشك من إصابة الطفل بمتلازمة دي جورج عندما يكون هناك مجموعة الأعراض المذكورة سابقاً، خاصة العيوب الخلقية في القلب - انقطاع في الشريان الأورطي، وجود شق في سقف الحلق، نقص مستوى الكالسيوم بالدم، وغيرها، حيث يقوم بإجراء العديد من الاختبارات والفحوص ومنها: o القصة المرضية للعائلة لمعرفة وجود حالات مشابهة o القصة المرضية للحمل o قياس مستوى هرمون الغدة جار الدرقية parathyroid hormone o قياس مستوى الكالسيوم والفسفور والإنزيم القلوي الفسيفتائيAlkaline Phosphatase Enzymes o أشعة سينية للصدر - توضح غياب الغدة السعترية o نسبة كريات الدم البيضاء الليمفاوية o اختبارات لقياس جهاز المناعة(مجموع الكريات الدم البيضاء الليمفاوية من نوع بي وتي) CD3+/CD4+ o قياس الأجسام المضادة عند الضرورة o أشعة صوتية للقلب (echocardiography) o تخطيط القلب o فحص الكروموسومات o فحص الكروموسومات باستخدام تقنية تهجين الفلوريسنت Fluorescent In Situ Hybridization .jpg) المضاعفات المرضية والعلاج: لا يوجد علاج لحالات متلازمة دي جورج، لأن المشكلة خلقية وموجودة في جميع خلايا الجسم، ولكن هناك طرق وعلاج لبعض المشاكل المرضية المصاحبة، يمكن من خلال التعرف عليها في وقت مبكر السيطرة عليها وتقليل مخاطرها على الطفل، والطبيب المعالج هو من يقدر تلك الحالات وكيفية التعامل معها، ومنها: o مشاكل القلب: أغلب الأطفال المصابين يكون لديهم مشاكل في تكوين القلب، تتراوح خطورتها من مشاكل بسيطة إلى مشاكل تؤدي للوفاة، لذلك فقد يستدعي الأمر تدخل جراحي بشكل مبكر بعد الولادة مباشرة لتصحيح العيوب القلبية، وذلك حسب نوع المشكلة. o فقدان المناعة: لم تنجح حتى الآن حالات زراعة الغدة السعترية أو خلايا العظام للمصابين بهذه الحالة، لذى فإنه في حال وجود نقص للمناعة، فإن الطبيب المعالج يقوم بمحاولات لتخيف من تبعاتها، ومنها: 1. إذا كان الطفل يحتاج لجراحة القلب وفي حاجة إلى نقل دم، يجب أن يعالج الدم بالإشعاع قبل أن يعطى للطفل 2. يجب أن يخضع هؤلاء الأطفال إلى إعادة تقييم مناعتهم قبل إعطائهم التطعيم الثلاثي البكتيري MMR ، والتطعيم ضد العنقز - الجديري المائي o الكالسيوم المنخفض: في حال وجود انخفاض مستوى الكالسيوم في الدم، يقوم الطبيب المعالج بإضافة الكالسيوم للغذاء، كما يقوم بإضافة فيتامين دال - ون ألفا One Alpha ـ الذي يعمل على تصحيح مستوى الكالسيوم ويقلل الفوسفات o الترجيع والقيء والصعوبة في الأكل الأطفال المصابون بالشفة الأرنبية وشق الحنك، كما صغر حجم الحنك، يلاحظ لديهم صعوبة في المص والمضغ والبلع، كما زيادة نسبة حدوث الترجيع والقيء من الأنف، مما يؤثر على صحتهم ونموهم، مما يستدعي تدخل أخصائي البلع والتغذية، واحتمالية استخدام التغذية عن طريق الأنبوب لمدة من الزمن. o النطق والتخاطب: الأطفال المصابون بالشفة الأرنبية وشق الحنك يكون لديهم مشاكل في النطق والتخاطب، مما يستدعي التدخل الجراحي، كما أخصائي النطق. o مشكلة السمع: التهابات الأذن الوسطى مشكلة شائعة، وذلك يجب متابعة الحالة لتفادي فقدان السمع o مشاكل التواصل: هذه المشكلة يمكن أن يكون سببها مشكلات تتعلق بالسمع والتخاطب، أو ربما تكون لها علاقة باستيعاب اللغة مشاكل الكلى: يلاحظ زيادة العيوب الخلقية في الجهاز البولي، كما زيادة حدوث التهابات المسالك البولية، وتلك تحتاج لمتابعة وتدخل طبي o المشاكل النفسية: يلاحظ في الغالب أن للأطفال تقدير ذاتي منخفض وفاقدي الثقة، كما يلاحظ أنهم يكونوا بشكل أفضل عندما يكونوا مع شخص أكبر منهم ومألوف لديهم ويثقون فيه على خلاف ما يكونوا عليه عندما يكونوا مع أقرانهم. يجب ملاحظة أن هؤلاء الأطفال في الغالب يعانون من "قلة التركيز والانتباه" ، وفي هذه الحالة يجب عدم إعطاءهم أدوية مثل ريتالين Ritalin ذلك بأن هذا الدواء قد يحدث آثاراً عكسية . في الغالب يعاني المرضى من مشكلات صحية ذهنية مثل الكآبة ذات القطبين أو الكآبة المهووسة، يجب استشارة أخصائي في حالة الشك بحدوث مثل هذه المشاكل. o التأخر في التعليم : معظم الأطفال المصابون لديهم مشكلات تعليمية دنيا أو طفيفة، يحتاجون إلى بعض المساعدة داخل فصول الدراسة، كما يحتاجون على اهتمام على المدى البعيد للتأثير الايجابي على حاجاته الصحية مآل المرض: o المشكلة خلقية، وفي جميع الخلايا، ولا يتوقع لها الشفاء الذاتي o لا يوجد علاج طبي أو جراحي o درجة المرض والأعراض تختلف من طفل لآخر o تصل نسبة حدوث الوفاة خلال السنة الأولى إلى 80%، وخاصة مع وجود العيوب القلبية .jpg) التطعيم ومتلازمة دي جورج: الهدف من التطعيم هو تعزيز جهاز المناعة، ففي حالة وجود البكتريا أو الفيروس يمكن لجهاز المناعة التفاعل بسرعة وفعالية، ومن خلال أعطاء التطعيم يمكن لنا بناء المناعة لدى الأطفال الطبيعيين، وتنقسم التطعيمات إلى قسمين: 1. التطعيمات غير النشطة: وهي التي يتم إعدادها من جزيئيات من البكتريا ، الفيروس الميت، مثل السعال الديكي والالتهاب السحائي 2. التطعيم الحية المضعفة: وهي التي تحتوي على الجرثومة التي لا تسبب المرض في الأشخاص ذوي الجهاز المناعي الجيد، مثل السل، الحصبة الأطفال المصابون بمتلازمة دي جورج، ومع ضمور الغدة السعتية المسئولة عن المناعة، فإن التطعيمات يجب الانتباه لها وتعطى عن طريق متخصص، وتتركز في ما يلي: o الثلاثي الفيروسي DPT يعطى بأمان o هيموفلس أنفلونزا Hib يعطى بأمان o تطعيم السل BCG، الثلاثي البكتيري MMR ، العنقز Varicella يعطى بعد قياس مستويات الأجسام المضادة، وعدد خلايا تي T-cells o تطعيم شلل الأطفال الفموي Oral polio، يتم استبداله بشلل الأطفال العضلي intramuscular polio- Sabin العنقز - الجديري المائي: الإصابة بهذا المرض تحتاج إلى ذكر خاص لدى المصابين بمتلازمة دي جورج، والمصابين بنقص المناعة، فإذا كانت الحالة بسيطة في الأطفال العاديين، فقد تكون قاتلة لمن لديهم نقص مناعة، لذى ننصح بالابتعاد عن الأطفال المصابين، كما سرعة زيارة الطبيب لإعطاء جرعة من مصل أجسام مضادة للجديري المائي المضاد الحيوي: ربما تستدعي الحاجة إلى اخذ مضادات حيوية بصورة منتظمة لمنع الإصابات حتى يعمل جهاز المناعة، ويوصي بعض الأطباء يأخذ المرضى مضاد حيوي باستمرار (سبترين أو باكتريم) ريثما يتم التعرف على نتائج الفحوصات، ففي حالة أن تكون لدى الطفل مناعة منخفضة يجب أن يعطى مضادات حيوية لإصابات البكتريا حتى لا تسؤ حالته . الأطفال والأسنان: عندما يفقد الأطفال أسنانهم أو أن يقابلوا طبيب أسنان، يعطى المريض في هذه الحالة مضاداً حيوياً ليمنع الإصابة . ويكون هذا في غاية الأهمية في حالة وجود مشاكل بالقلب تؤدي إلى زيادة المخاطرة لدى الطفل لإمكانية تعرضه بالتهاب بطانة القلب |
|
|
|
|
| مواقع النشر (المفضلة) |
| الذين يشاهدون محتوى الموضوع الآن : 1 ( الأعضاء 0 والزوار 1) | |
|
|
 المواضيع المتشابهه
المواضيع المتشابهه
|
||||
| الموضوع | كاتب الموضوع | المنتدى | مشاركات | آخر مشاركة |
| جميل يستحق كل جميل | زمن الطوفان | الساحة الرياضيّة | 5 | 03-31-2011 07:15 AM |
| هدف جميل ... | & naif & | المقاطع الرياضية المرئية | 6 | 11-26-2009 05:31 AM |
| كيفية تعليم الطلاب للإعراب جميل جميل جدا جدا جدا جدا | الرهيب معكم | الملفات والفلاشات والبرآمج التعليميه | 1 | 09-06-2009 12:49 PM |
| د عاء جميل | سهيل الجنوبي | مكافحة البدع والمواضيع المشبوهة | 4 | 05-13-2007 01:34 PM |
| جميل المحيا | جبل حرفه | عطر الكلمات | 0 | 03-07-2004 12:29 AM |






 العرض العادي
العرض العادي
